DrTarget uses SOLACE
Structured Organization of Layers with Assay Classification and Evaluation
A proprietary ensemble of machine learning algorithms
fed by a hierarchical assay classification and scoring routines.
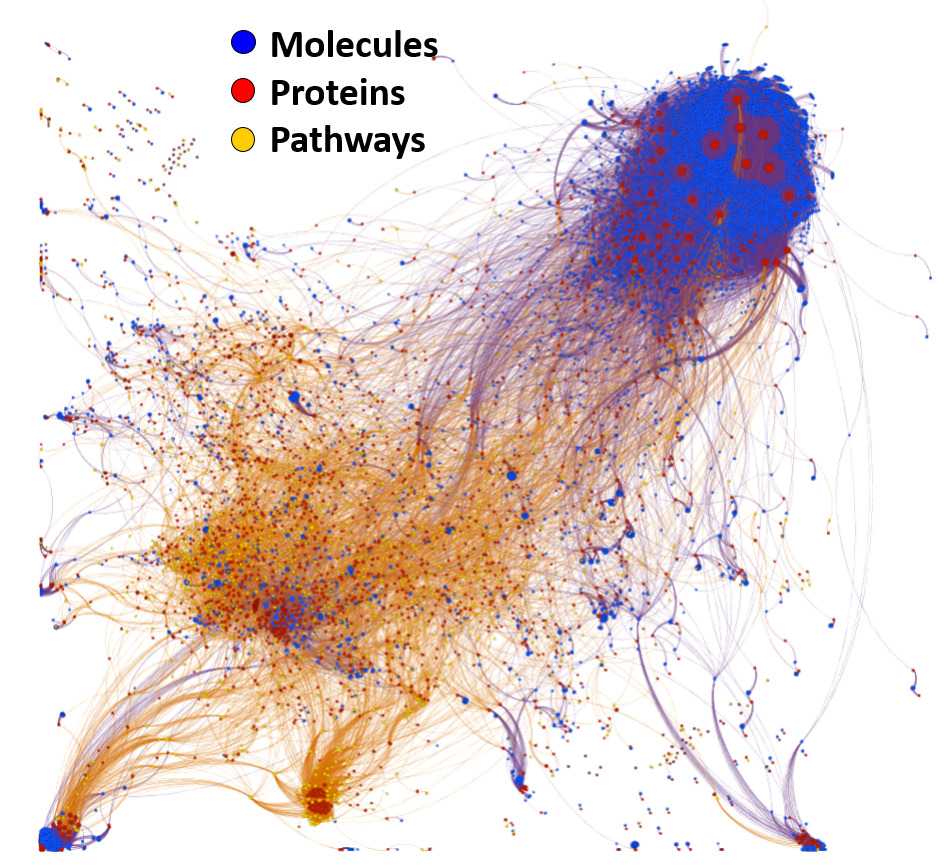
molecule-gene-pathway interaction map for predicted active molecules
DrTarget uses artificial intelligence to conduct virtual screens based on phenotypic descriptors of physiological states/diseases on ChEMBL DB, resulting in the prediction of the activity of 1.5M ChEMBL molecules on the selected experimental models.
Machine learning models employed are mainly based on interactions between molecules, assays, proteins, cell/tissues/animals, pathways and diseases, producing interaction network maps that can be aggregated at different levels to provide scores at the molecule, protein/gene and pathway level.
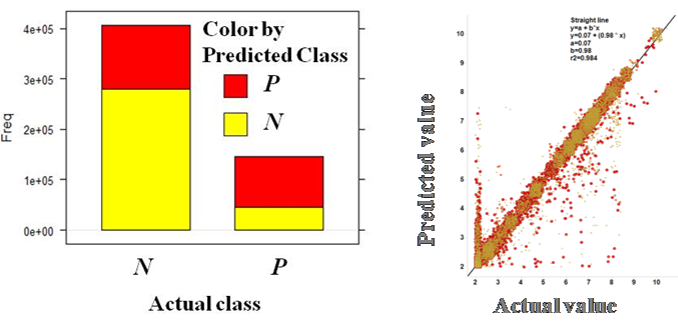 What if we wanted to build a focused, ad hoc, cheap compound collection for a particular experimental model or phenotype? We could use the recorded activities upon the target of our choice and look at what these molecules have done in the rest of the assays present in the DB, and then, use neuronal networks, random forests or many other machine learning tools to build a model through which we can pass molecules that have never seen our target to predict its activity. All the models incorporate measures of accuracy, precision, estimations of the goodness of the model and true and false positive rates. The page contains several examples of predictive analysis applied to different experimental cases taken from the ChEMBL downloadable information.
What if we wanted to build a focused, ad hoc, cheap compound collection for a particular experimental model or phenotype? We could use the recorded activities upon the target of our choice and look at what these molecules have done in the rest of the assays present in the DB, and then, use neuronal networks, random forests or many other machine learning tools to build a model through which we can pass molecules that have never seen our target to predict its activity. All the models incorporate measures of accuracy, precision, estimations of the goodness of the model and true and false positive rates. The page contains several examples of predictive analysis applied to different experimental cases taken from the ChEMBL downloadable information.
additional machine learning studies
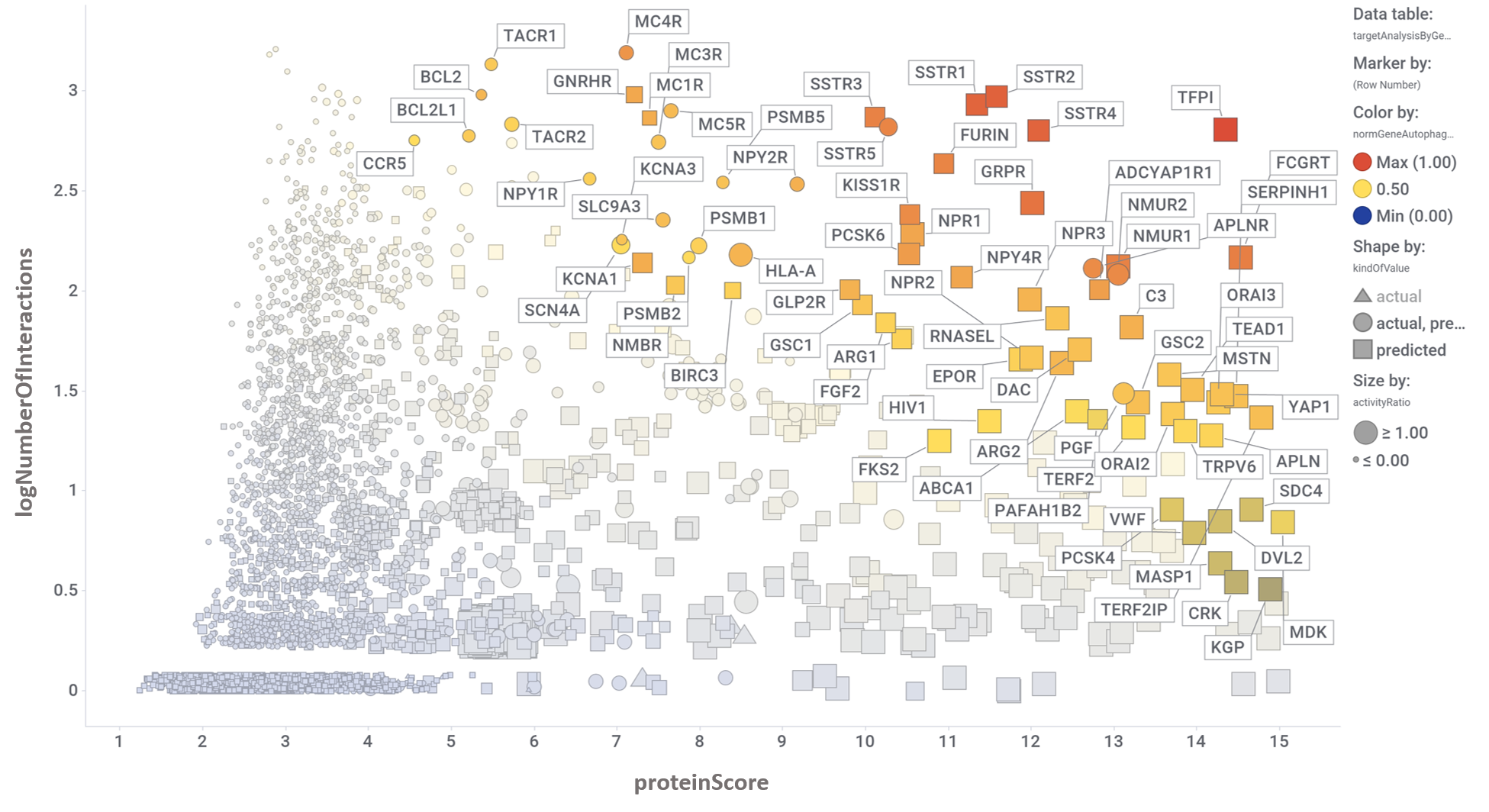
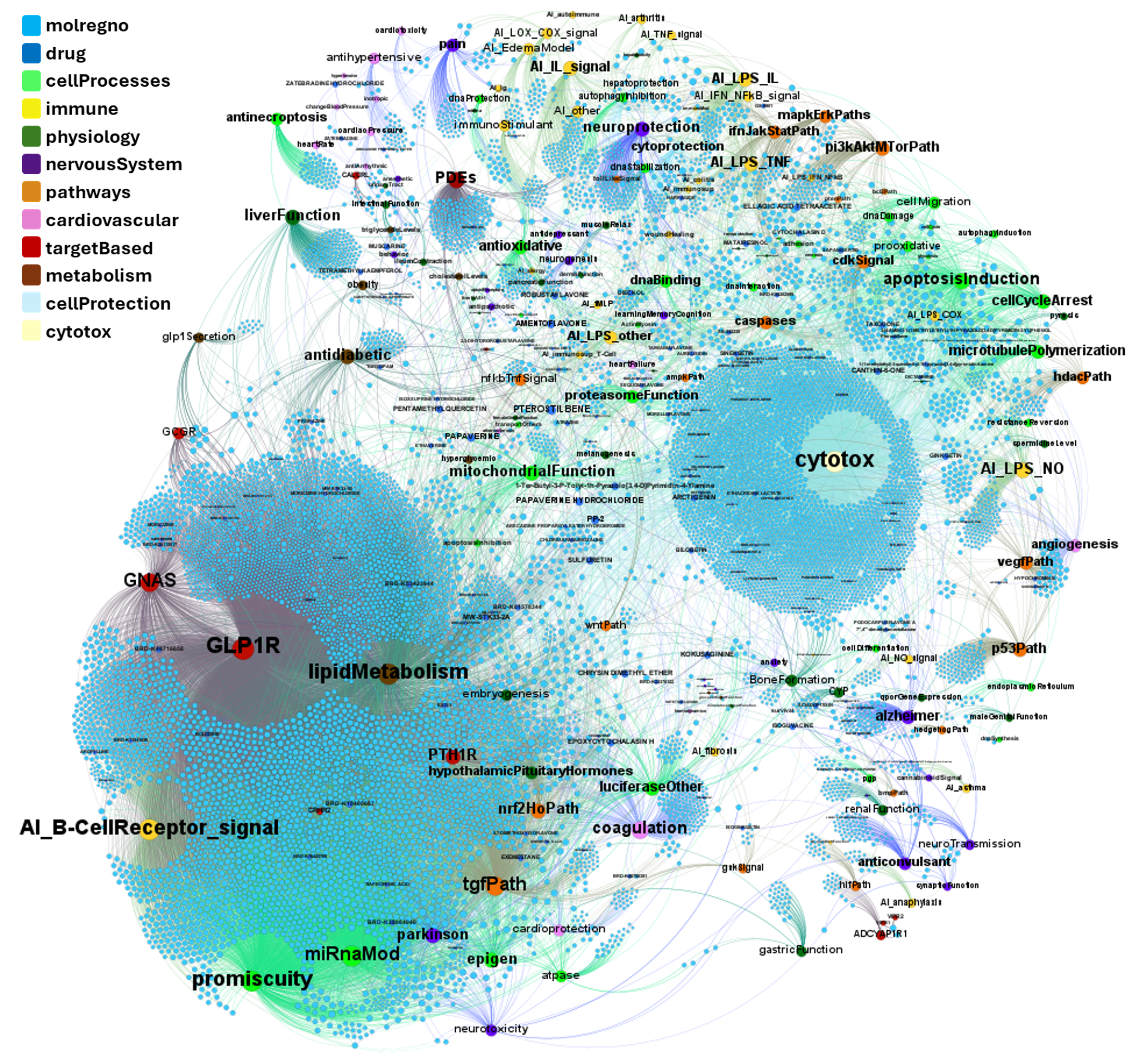
The complete interactions map of the predicted active molecules can help us to understand which are the relevant players in our model.
By aggregation of activity predictions at the protein/gene level, we can obtain activity scores based on the quality and number of interactions, thus identifying most relevant targets.
Section is filled with the examples below.
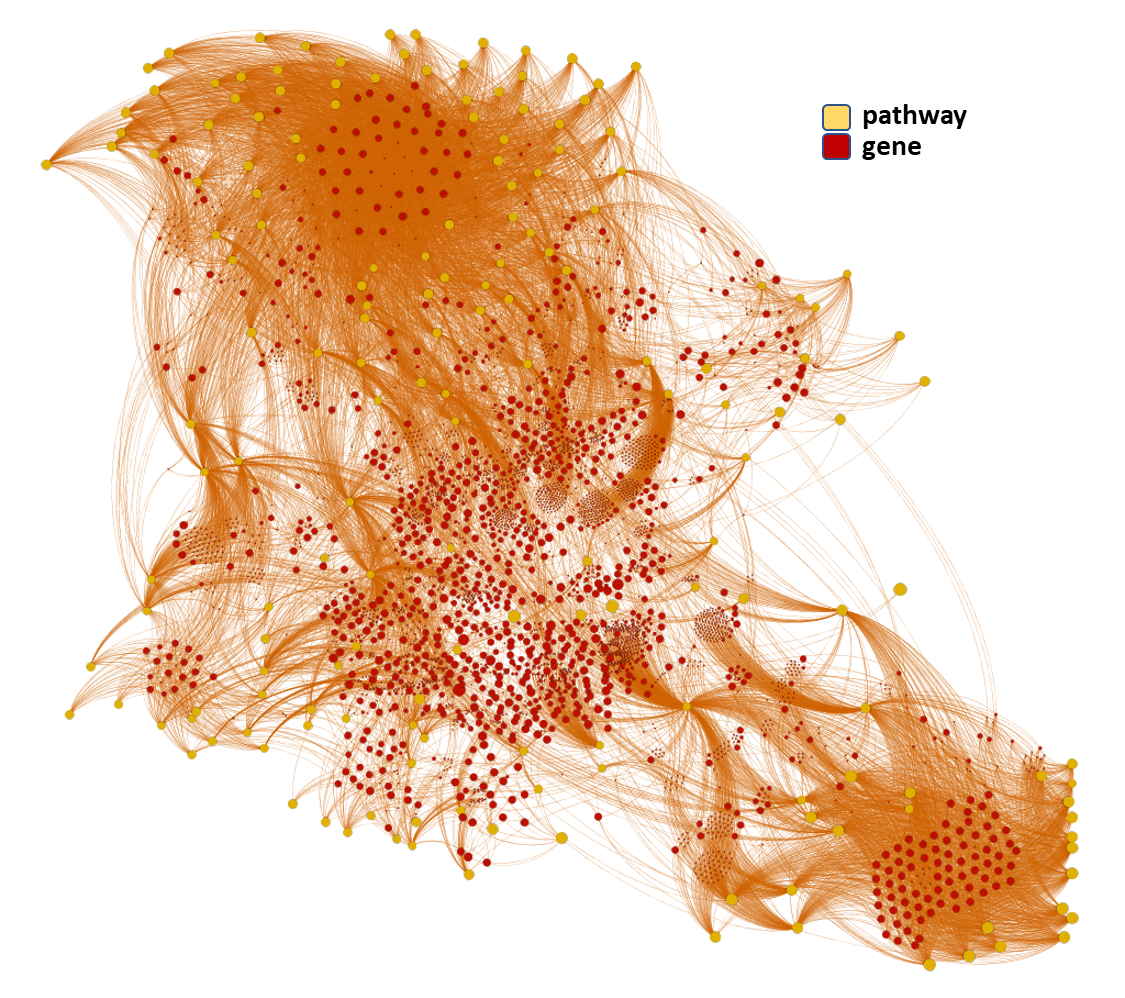
Here, we’ll focus on pathways-protein/gene networks.
The databases described in DBs section have incorporated pathway info from Reactome repositories summarizing the interaction of proteins in networks belonging to different biosystems, tissues and species.
Millions of records to be used for the detection of key biological cascades involved in diseases, protein targets assessment or identification of new proteins beyond ChEMBL records for being part of relevant pathways.
Drug Repositioning
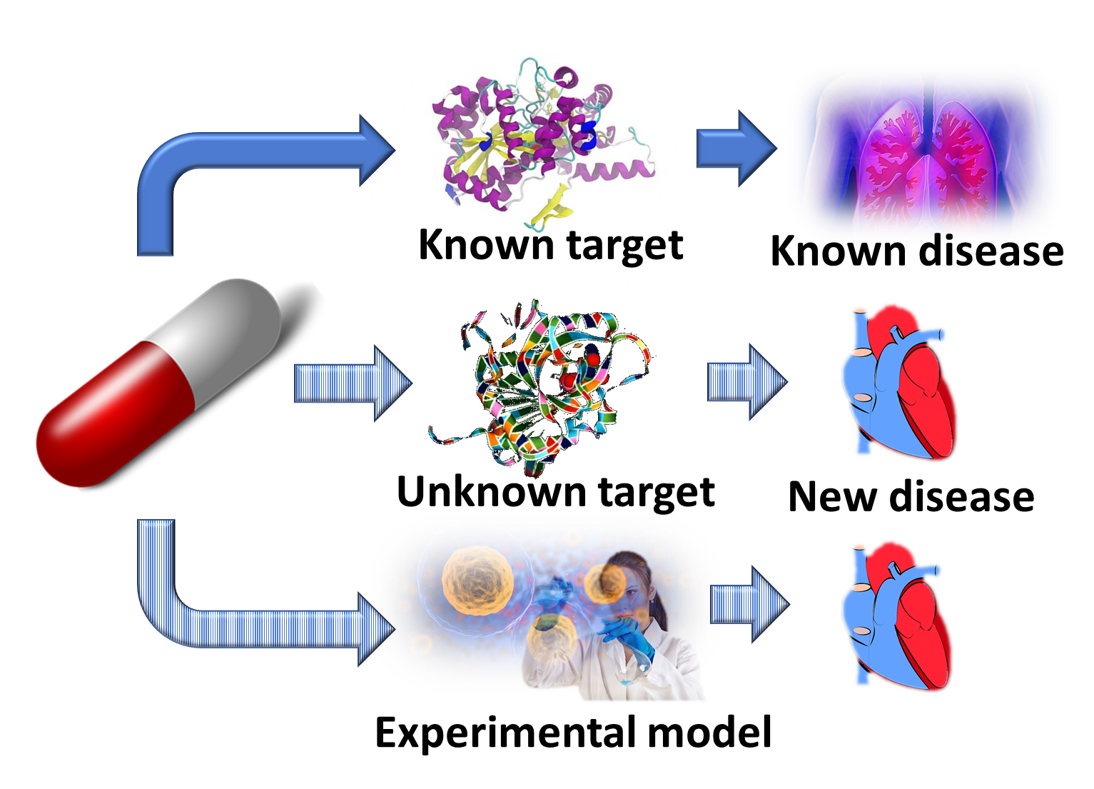
A significant advantage of drug repositioning over traditional drug development is that since the repositioned drug has already passed a significant number of toxicity and other tests, its safety is known and the risk of failure for reasons of adverse toxicology are reduced. More than 90% of drugs fail during development,[2] and this is the most significant reason for the high costs of pharmaceutical R&D. In addition, repurposed drugs can bypass much of the early cost and time needed to bring a drug to market. It significantly reduces the transition of bench research work to treatment at bedside. On the other hand, drug repositioning faces some challenges itself since the intellectual property issues surrounding the original drug may be complex and from a commercial point of view it may not always make sense to take such a drug to market.
Drug repositioning has been growing in importance in the last few years as an increasing number of drug development and pharmaceutical companies see their drug pipelines drying up and realize that many previously promising technologies have failed to deliver ‘as advertised’. Computational approaches based on virtual screening of comprehensive libraries of approved and other human use compounds against large numbers of protein targets simultaneously have been developed to enhance the efficiency and success rates of drug repositioning, particularly in terms of high-throughput shotgun repurposing.[3][4][5]
Drugs and molecules, gene expression, genes and pathways classification, nutraceuticals, phenotypic profiles, in vivo activity and compound-protein interactions from multiple DBs.

![]()
![]()
![]()
![]()
![]()
![]()

![]()
DrTarget methodology workflow, DBs & tools.
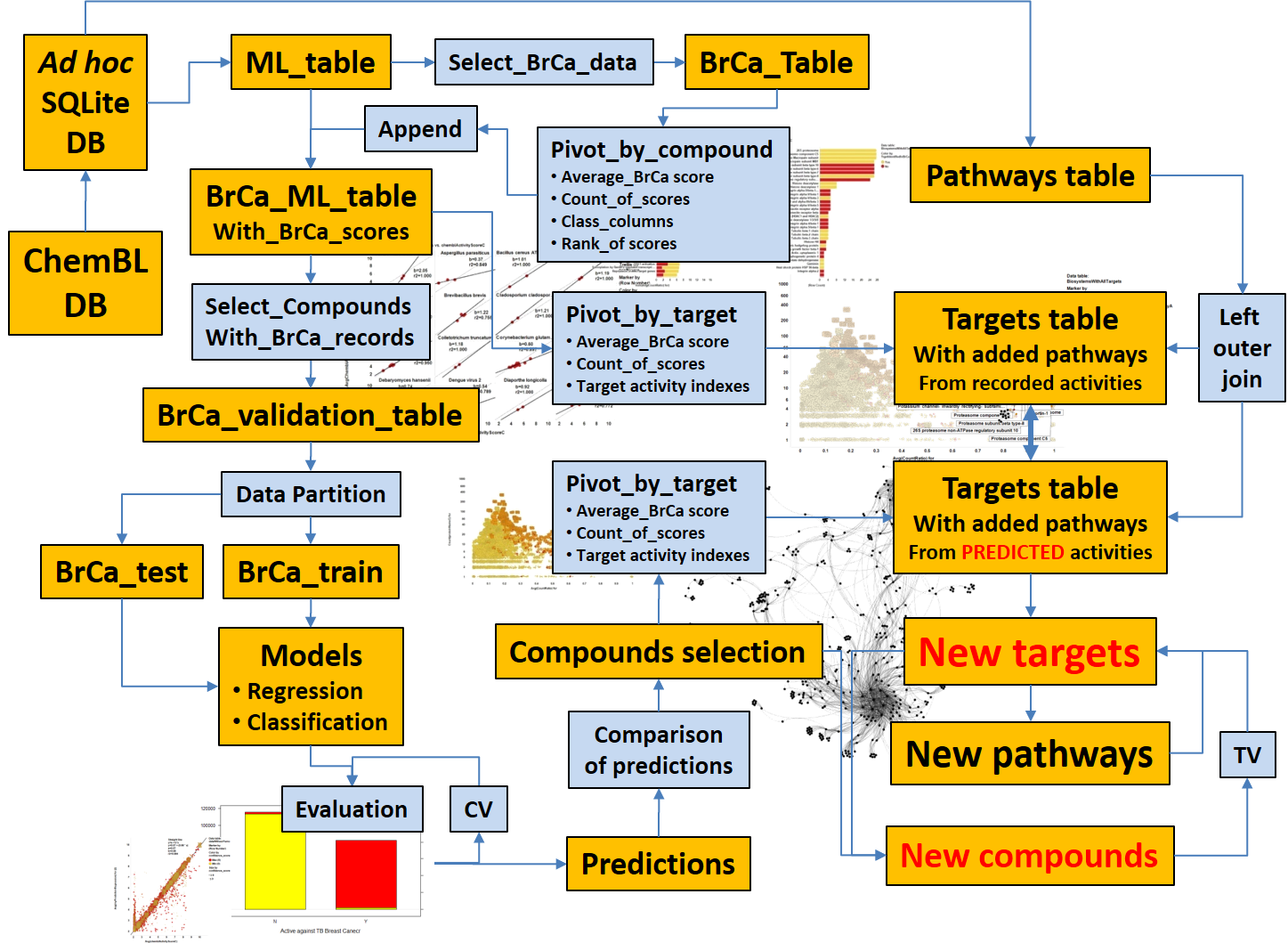
Starting from an appropriate data frame containing chemistry, biology and assay information, built from SQL querying onto the selected ML DB, we can develop prediction and identification procedures. This section summarizes the subsequent procedures and algorithms employed. Whatever the route we take to identify pathways or targets of interest, or putative bioactive molecules, we can make them converge to confirm each other’s predictions.
The schema on the left attempts to illustrate how methodology described in the links below sustains an integrative prediction modelling.