This site describes a Machine Learning effort that uses different public experimental repositories to conduct a virtual screening on 1.8M ChEMBL molecules that identifies 64k predicted active compounds against SARS coronaviruses. All molecular interactions of these compounds with proteins with experimental records in ChEMBL are then explored to discover potential mechanisms of action. The site contains links leading to detailed analysis of more specific network graphs, compound structures and literature references supporting most relevant findings.
Click on specific tabs to review SARS-CoV biology, data sources, methodology, Evaluation of Machine Learning models and final predictive outcome.
By We Are Covert – We Are Covert, Copyrighted free use
Severe acute respiratory syndrome coronavirus
Severe acute respiratory syndrome coronavirus (SARS-CoV or SARS-CoV-1)[2] is a strain of virus that causes severe acute respiratory syndrome (SARS).[3] It is an enveloped, positive-sense, single-stranded RNA virus which infects the epithelial cells within the lungs.[4] The virus enters the host cell by binding to angiotensin-converting enzyme 2.[5] It infects humans, bats, and palm civets.[6][7]
On 16 April 2003, following the outbreak of SARS in Asia and secondary cases elsewhere in the world, the World Health Organization (WHO) issued a press release stating that the coronavirus identified by a number of laboratories was the official cause of SARS. The Centers for Disease Control and Prevention (CDC) in the United States and National Microbiology Laboratory (NML) in Canada identified the SARS-CoV-1 genome in April 2003.[8][9] Scientists at Erasmus University in Rotterdam, the Netherlands, demonstrated that the SARS coronavirus fulfilled Koch’s postulates thereby confirming it as the causative agent. In the experiments, macaques infected with the virus developed the same symptoms as human SARS victims.[10]
A similar virus was discovered in January, 2020. This virus, named Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) is the causative pathogen of the ongoing COVID-19 pandemic.[11]
Virology
SARS-CoV-1 follows the replication strategy typical of the coronavirus subfamily. The primary human receptor of the virus is angiotensin-converting enzyme 2 (ACE2), first identified in 2003.[24]
Human SARS-CoV-1 appears to have had a complex history of recombination between ancestral coronaviruses that were hosted in several different animal groups.[25][26] In order for recombination to happen at least two SARS-CoV-1 genomes must be present in the same host cell. Recombination may occur during genome replication when the RNA polymerase switches from one template to another (copy choice recombination).[26]
SARS-CoV-1 is one of seven known coronaviruses to infect humans. The other six are[27]:
| Severe acute respiratory syndrome coronavirus | |
|---|---|
 | |
| Electron microscope image of SARS virion | |
| Virus classification | |
| (unranked): | Virus |
| Realm: | Riboviria |
| Kingdom: | Orthornavirae |
| Phylum: | Pisuviricota |
| Class: | Pisoniviricetes |
| Order: | Nidovirales |
| Family: | Coronaviridae |
| Genus: | Betacoronavirus |
| Subgenus: | Sarbecovirus |
| Species: | |
| Strain: | Severe acute respiratory syndrome coronavirus |
| Synonyms | |
| |
ChEMBL and PUBCHEM DBs store a good amount of records of molecules interacting with SARS proteases and inhibiting viral proliferation in diverse phenotypic assays carried out with different coronavirus species.
PubChem stores data from 2 different HTS efforts on more than 300k molecules:
Summary of probe development efforts to identify inhibitors of the SARS coronavirus 3C-like Protease (3CLPro) A direct measurement of viral protease activity.
qHTS of Yeast-based Assay for SARS-CoV PLP. A yeast reporter assay of protease activity.
ChEMBL, at this moment contains >7k data points from >200 different assays in 12 coronavirus species. Most of these datasets come from viral proliferation or cell death assays, although remains a small proportion of targeted models on proteases and polymerases.
And finally, a number of compounds reported to be active on coronavirus replication have been finally added to the model.
Data from described sources have been pooled together and a common SARS score has been generated based in their respective activities upon different assays. The common SARS score takes into account several activity records to configure a combined SARS score common for all stored assays (PCHEM score, pXC50, max inhibition, activity in secondary assays, area under the compound concentration-response curve -AUC- and others). The activities of several SARS protease inhibitors recently assayed for COVID-19 growth inhibition in cell cultures and humans and reported have also been included and a common SARS score has been calculated accordingly.

Example of parameters diversity on one of the SARS assays.
About 300k molecules have a record in at least one SARS assay, and more than 4M records in the ChEMBL DB associated to >130k assays carried out on 4700 different protein targets and 100k phenotypic assays.
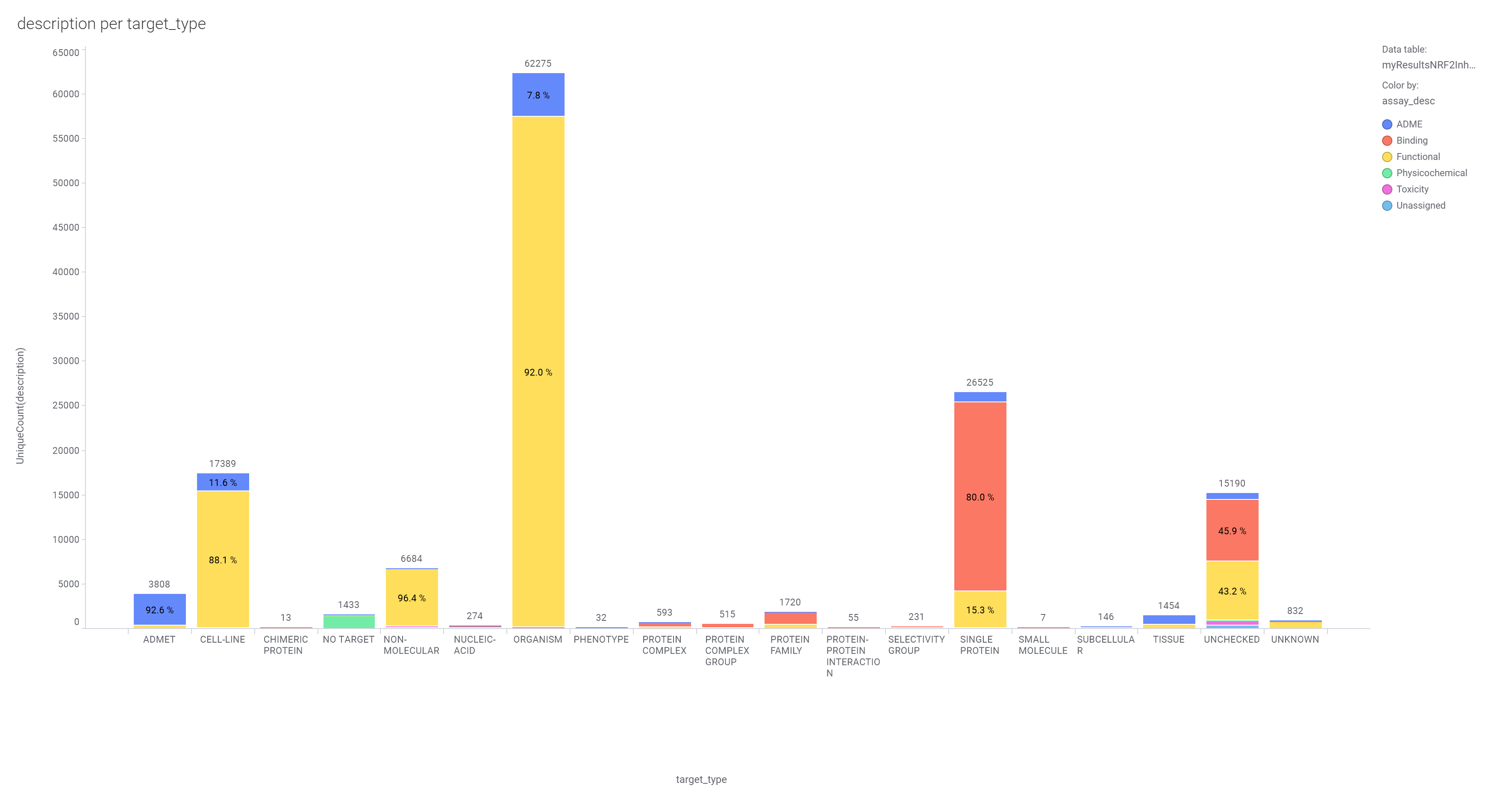
Assay diversity of the SARS data set.
The 4M ChEMBL records have been used to create a number of machine learning models that incorporate assay, organism, protein and molecular properties. Classification and regression random forests algorithms resulted the most successful among all. Classification algorithms return the active or inactive status for all the instances in the model. Results are aggregated at the molecule level creating a numerical value, countRatio, defined as the frequency with which a compound has been predicted active above a threshold.
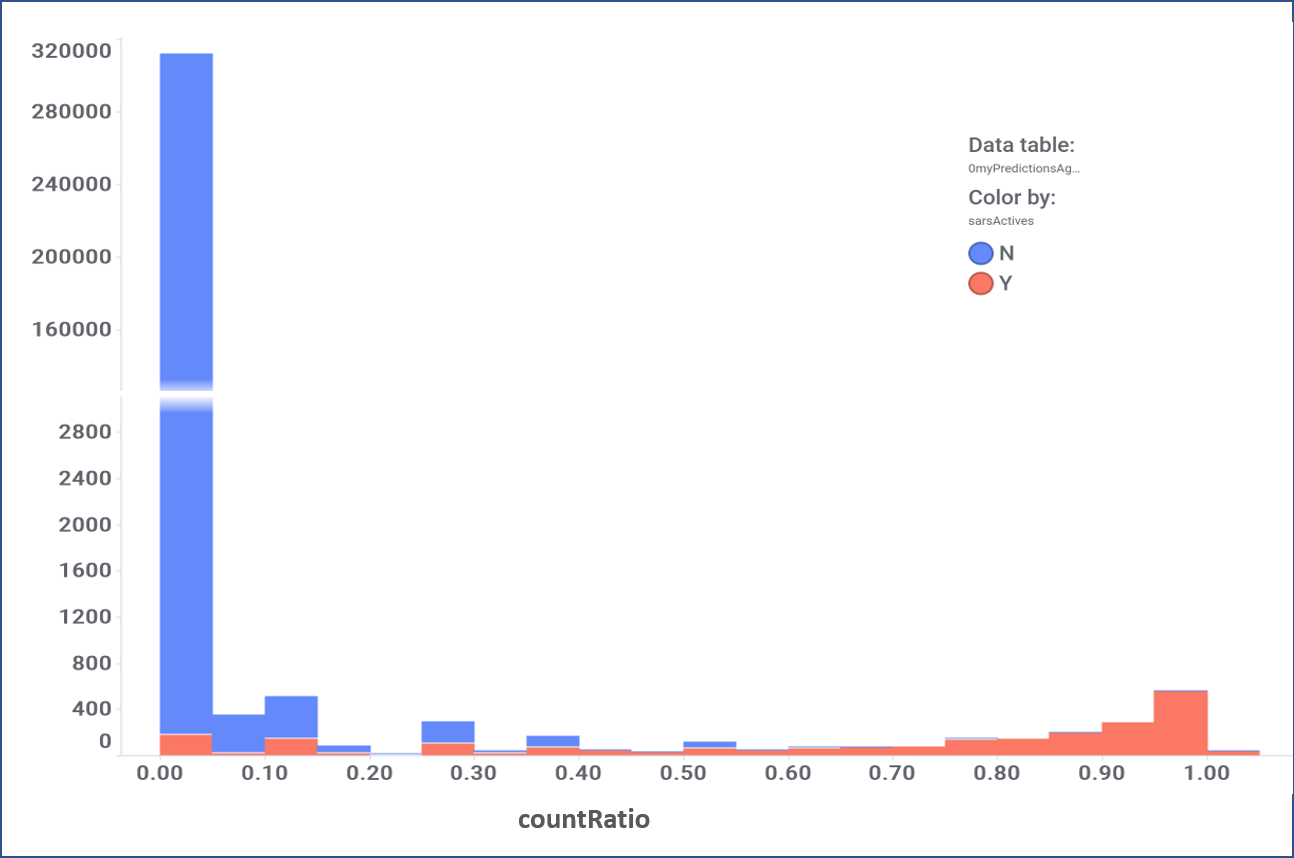
Evaluation of the performance of random forest classification. Color by actual label: red, active; blue, inactive.
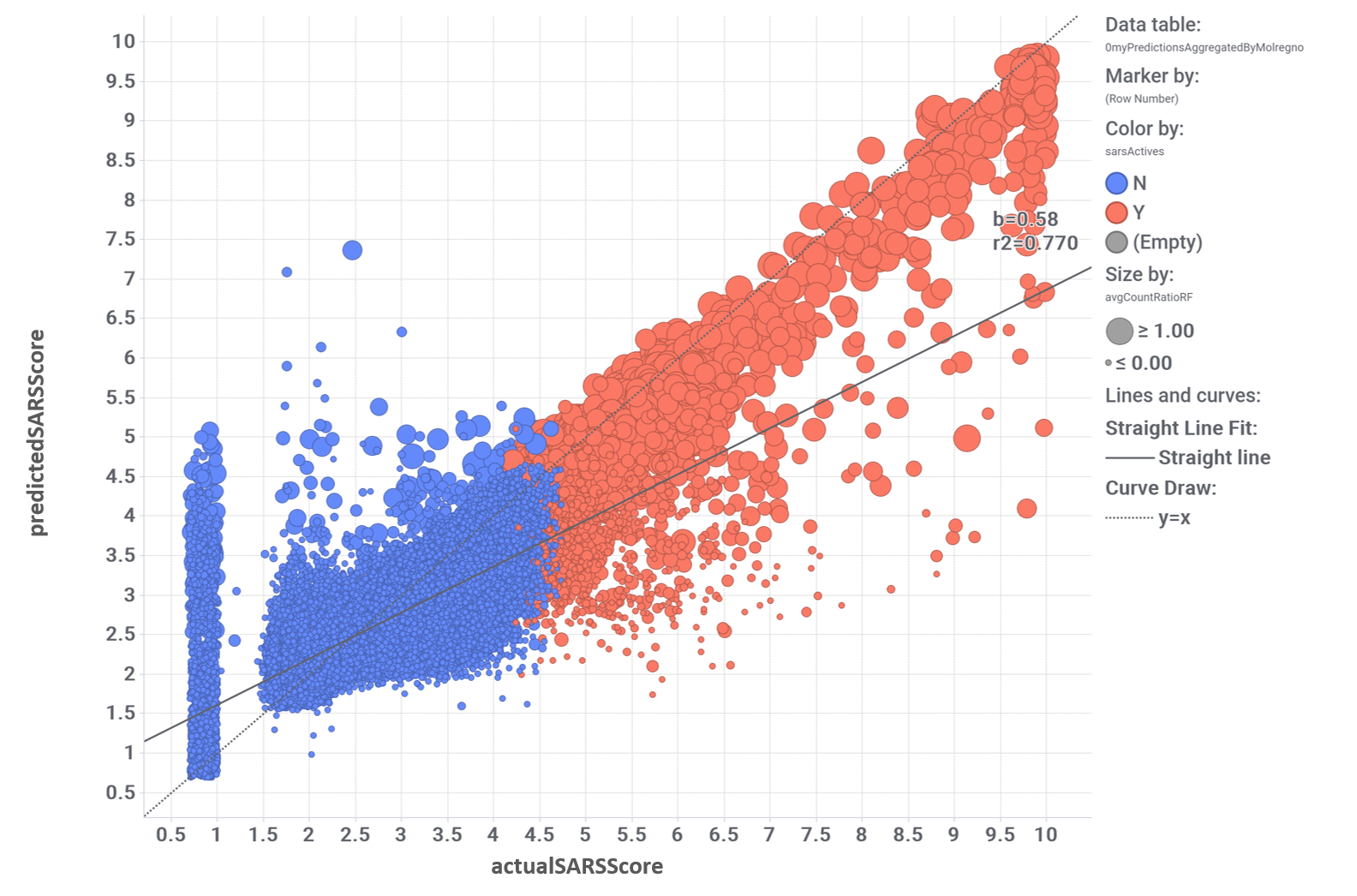
Evaluation of the performance of random forests regression algorithm. Actual vs predicted scores. Size by countRatio classification value. Color by actual label: red, active; blue, inactive.
Models validated as described in the “Machine Learning: Evaluation of predictive models” tab can be applied to the whole DB content, calculating a SARS predicted score for 1.8M compounds, and inally finding 64k active molecules for a 4.5 SARS score threshold.
Diagrams below showing the distribution of all active samples (left) and compounds similar to known SARS-CoV inhibitors described in literature (left).

Activity distribution histogram of the ChEMBL compounds.
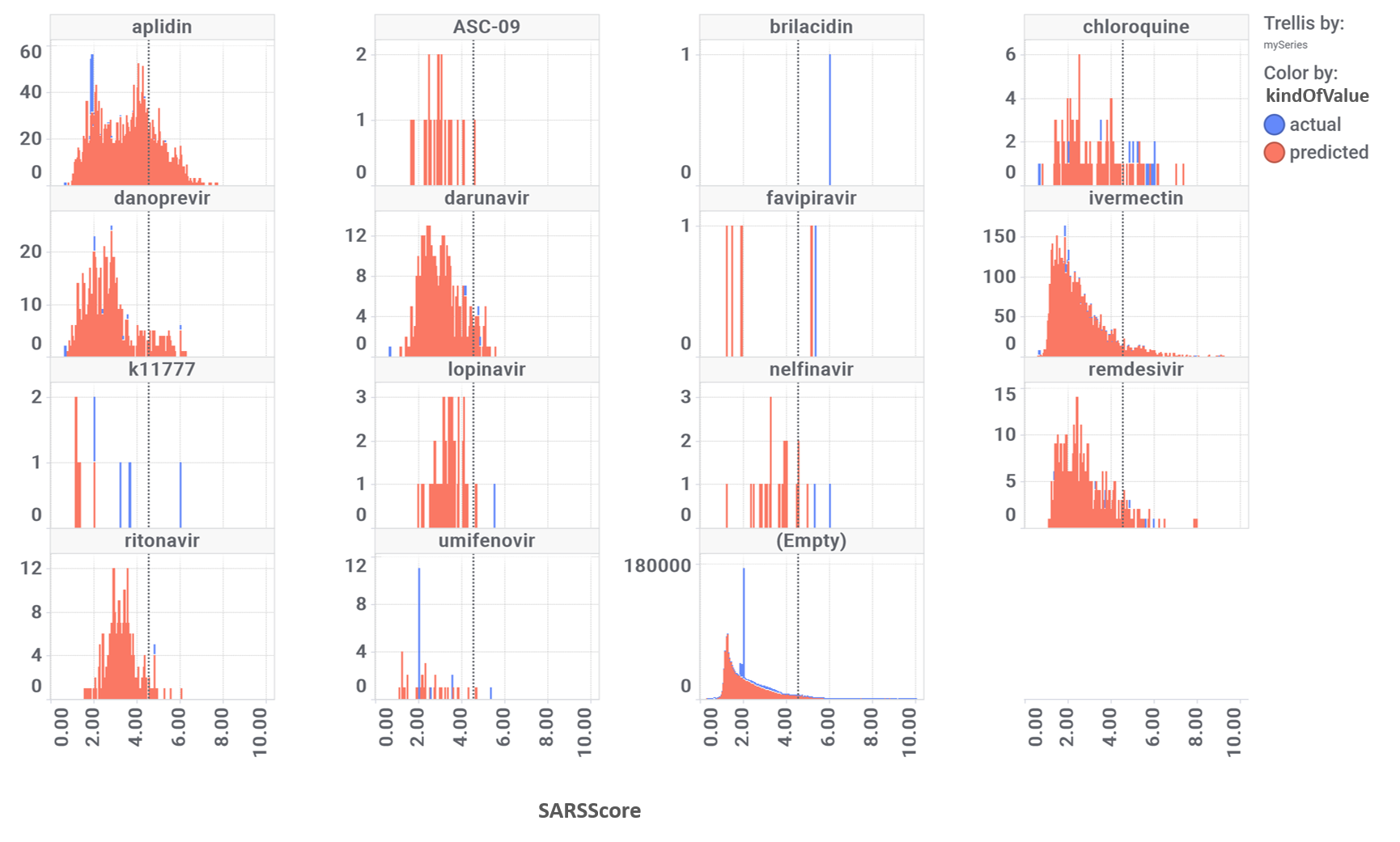
Activity distribution histogram of the known SARS inhibitors analogs. Click to expand image.
Now, we can interrogate ChEMBL DB to retrieve all interactions of the 64k molecules predicted as active with their target proteins and plot them in a network graph.
We can also add the pathway to which such proteins belong to unveil higher level dependencies.
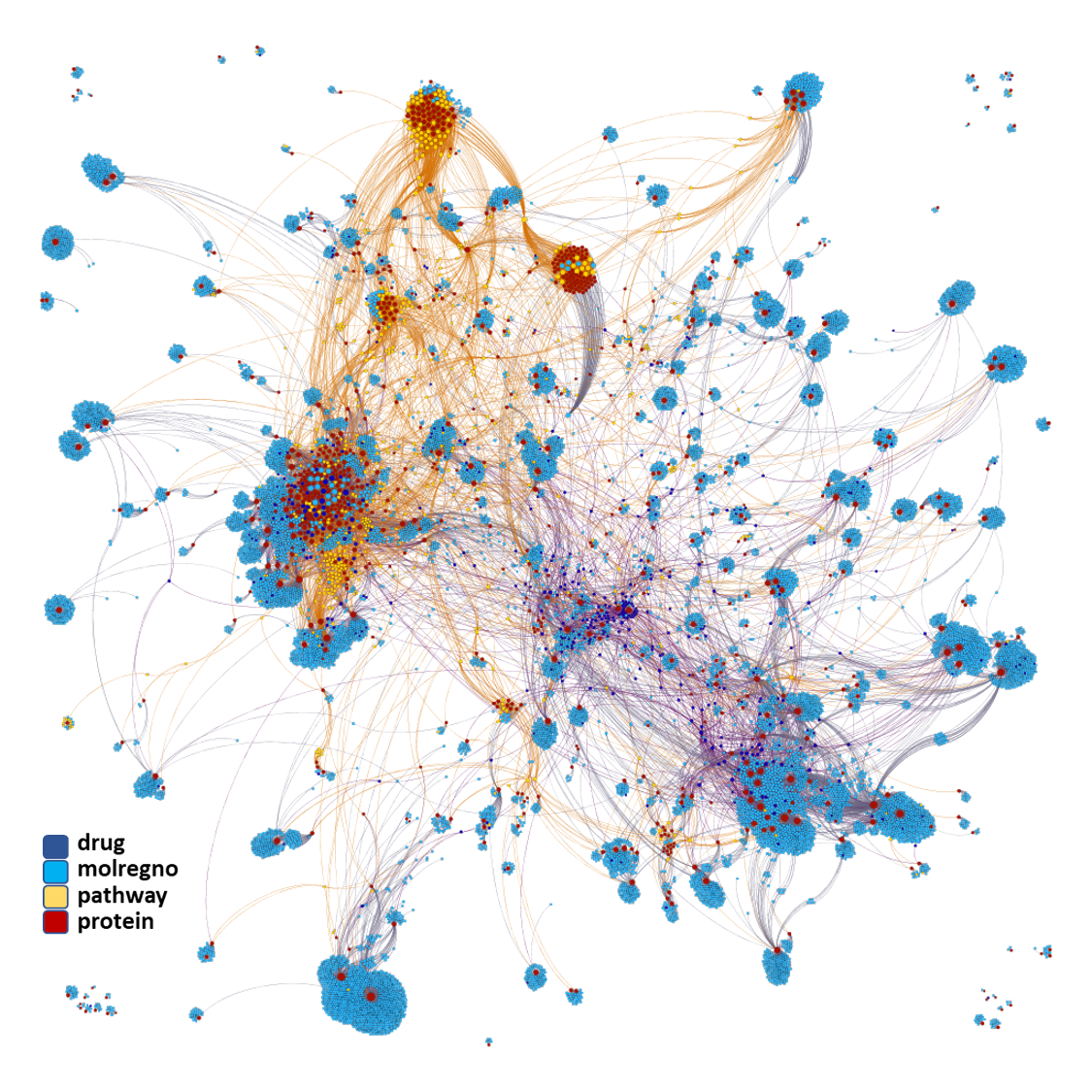
There is a lot of things we can do with our predicted active molecules.
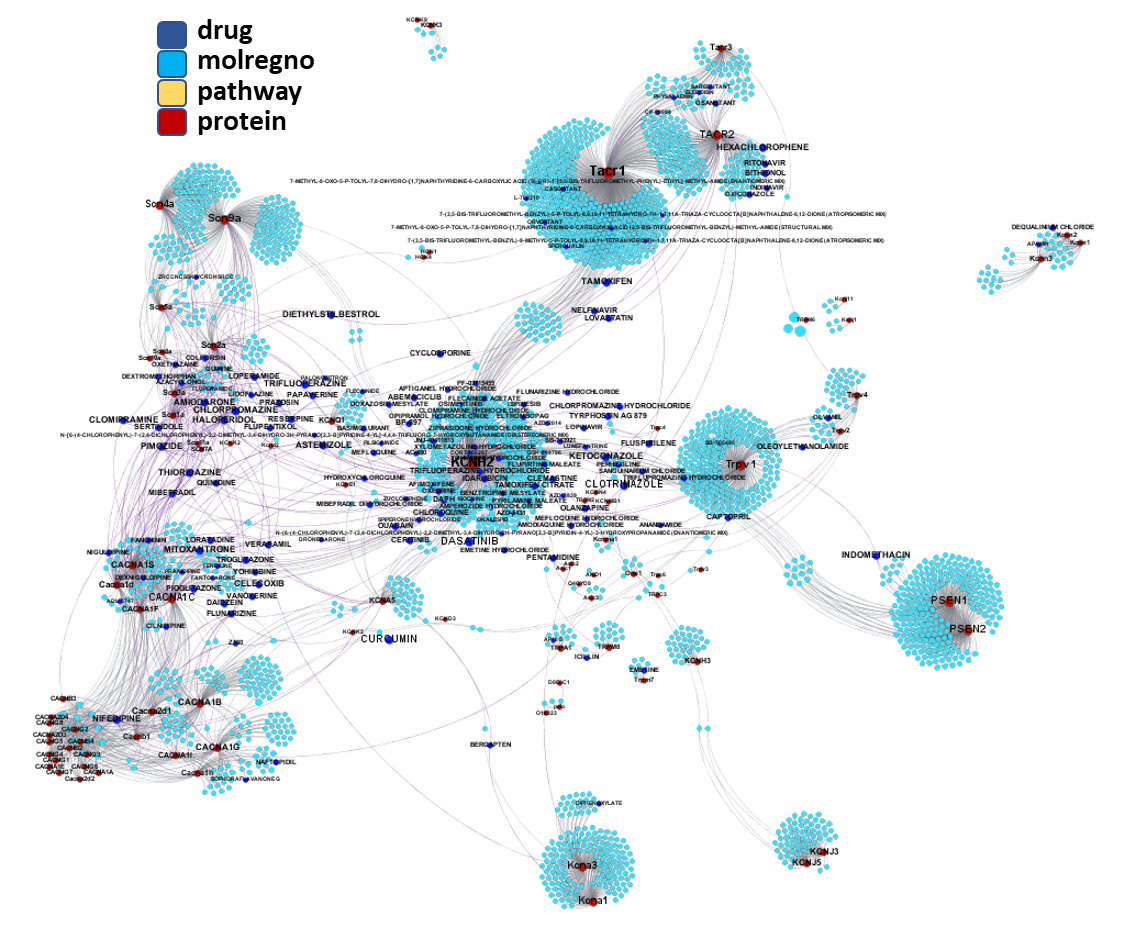
We can apply algorithms to map our molecule-protein-pathway interaction map into different quarters based on closest relationships, as you can see down left.
And we can classify all predicted active molecules on different clusters based on their structural similarity. The chart down right shows a dashboard where the scatter plot on the top represents the average SARS score for each cluster vs the number of molecules that compose it. The selection of particularclusters on the scatter plot determines the composition of the box plot below, illustrating the distribution of activity scores.

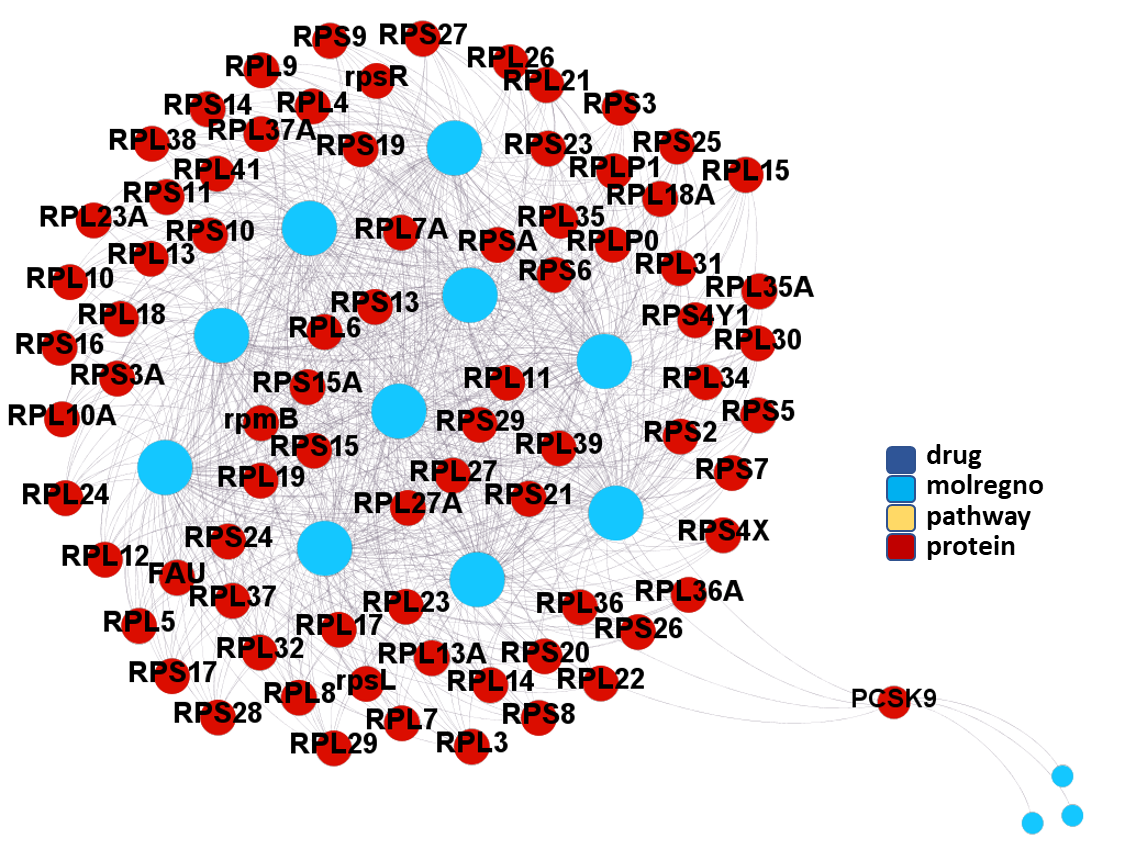
Modularity classification of the molecule-protein-pathway network graph obtained from predicted SARS active molecules interactions with proteins based on ChEMBL records. Each color is associated to a different module.
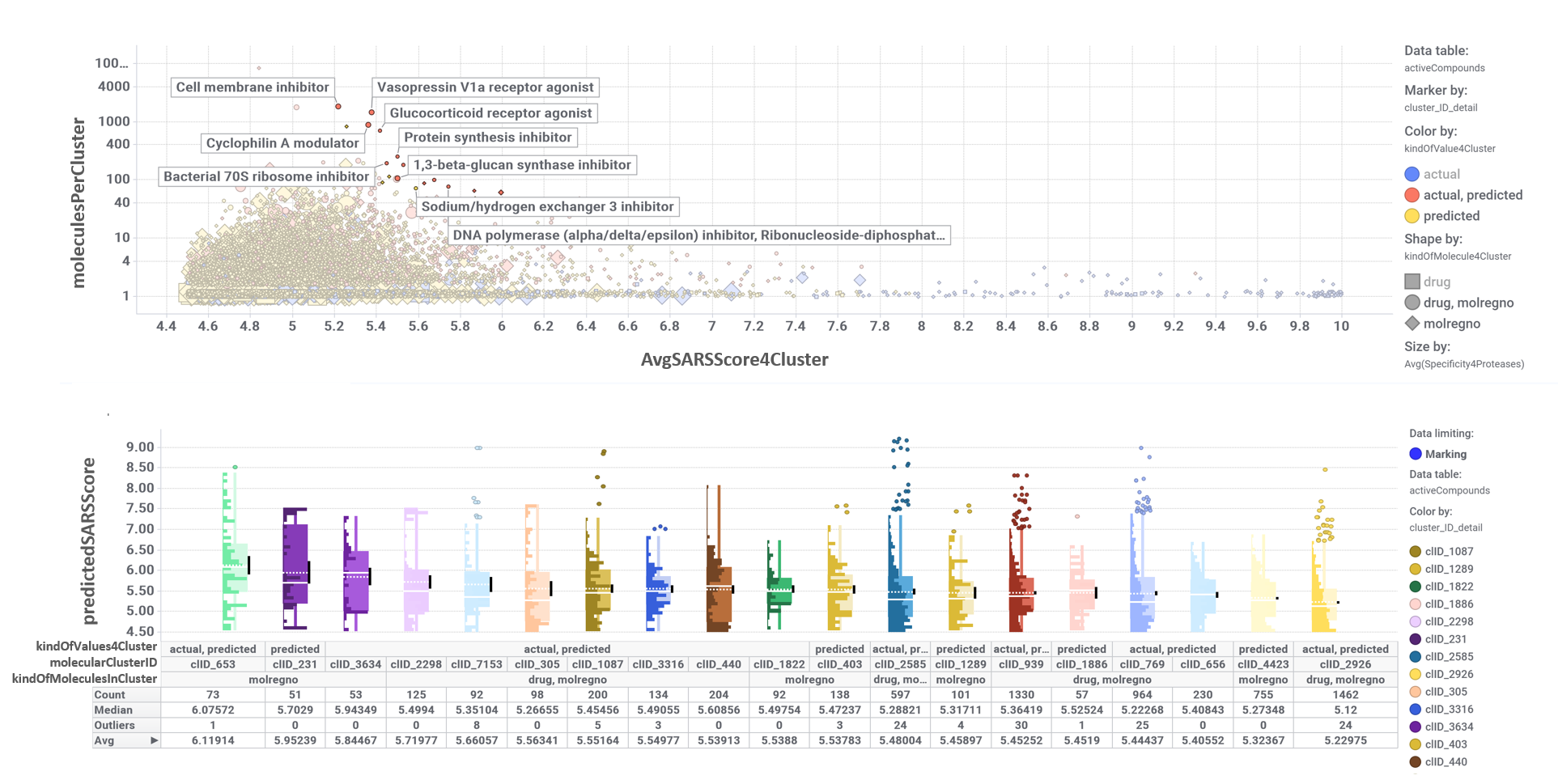
Activity distribution box plot of molecular clusters selected in the scatter plot by the mechanism of action recorded for some of their components.
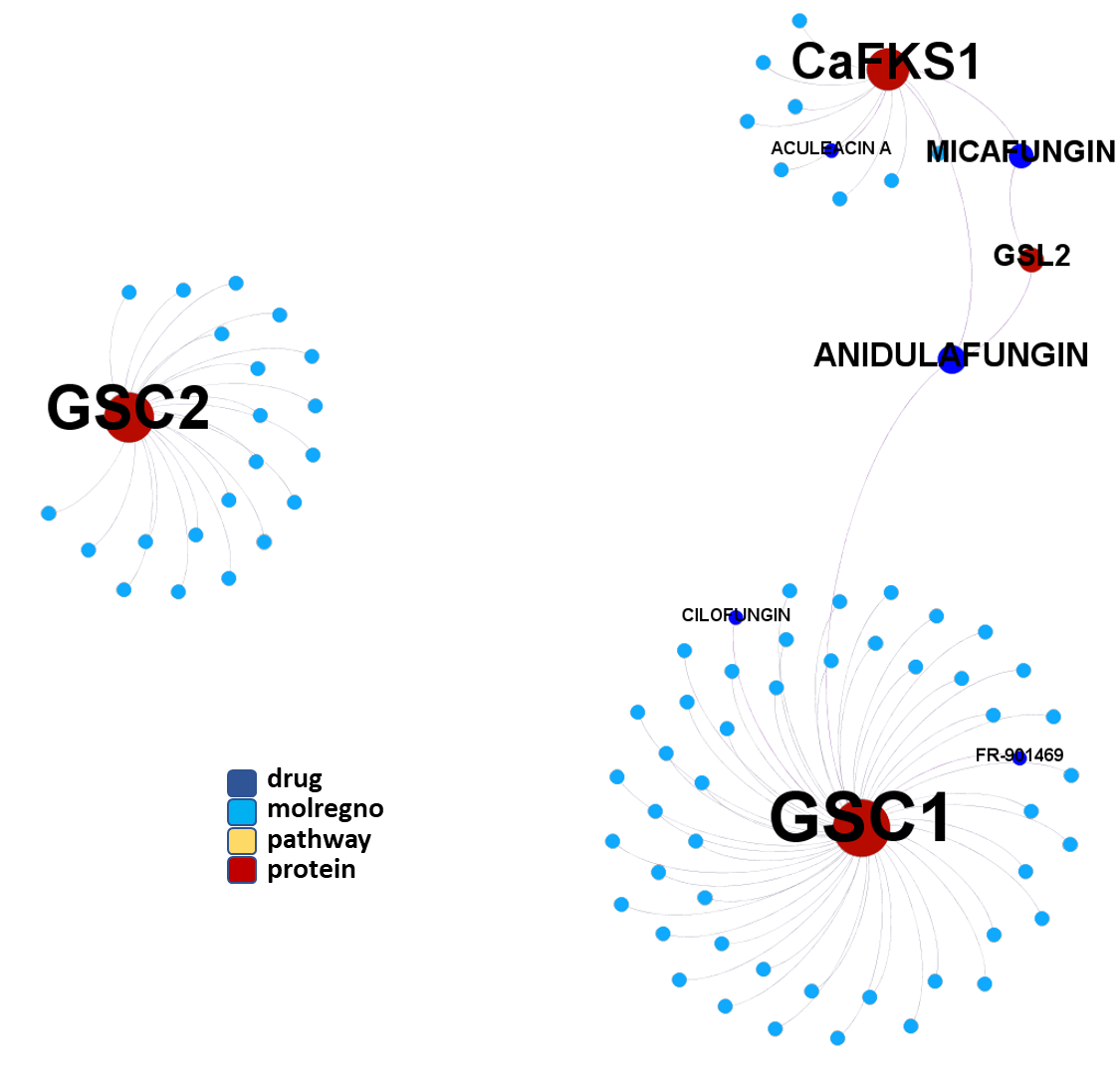
So, by gathering modularity criterion stablished from molecular interactions and compound clustering based on structural similarity, we can define sets of molecules associated to different mechanisms of action.
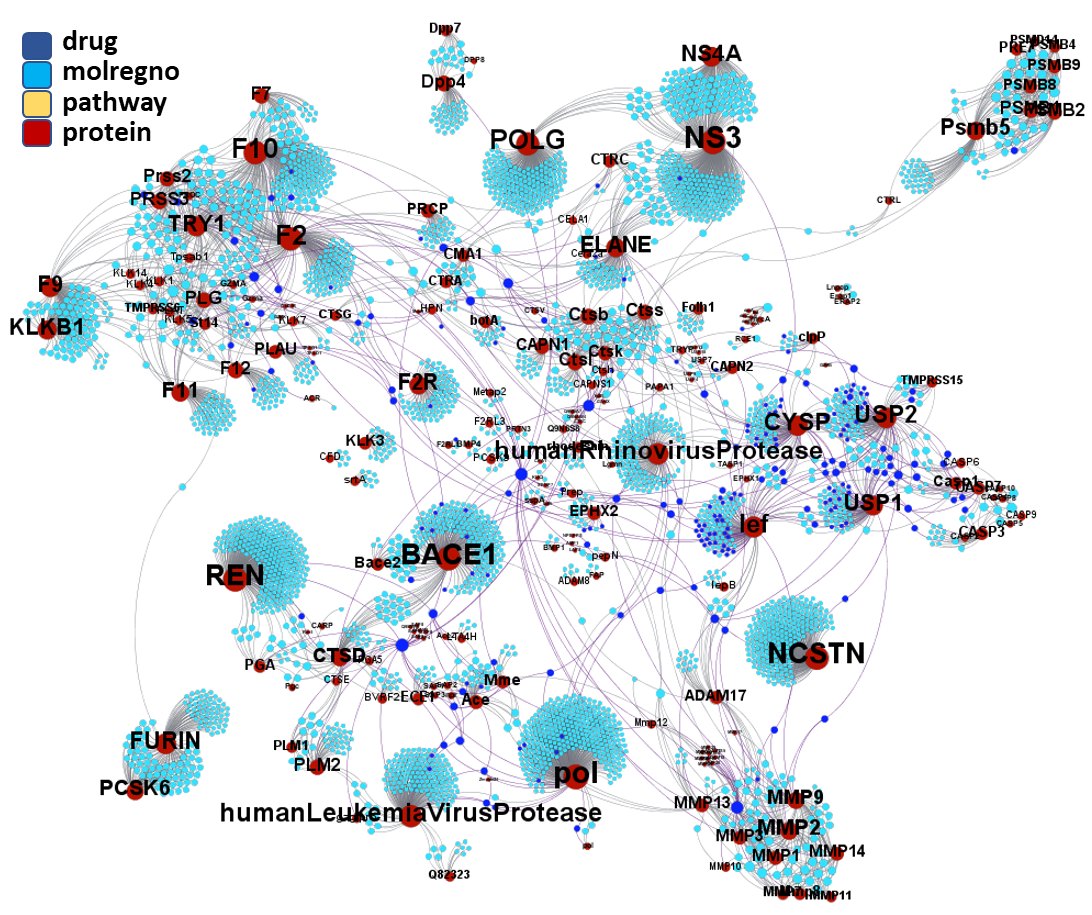
Click on the graphs below expand and visualize some of these sets, or start a longer trip clicking on the “Go to” buttons that will lead you to more specific interactions, compound structures and literature references supporting the potential role of compounds, targets or pathways as antiviral and, in many cases as anti SARS-CoV agent s or targets.
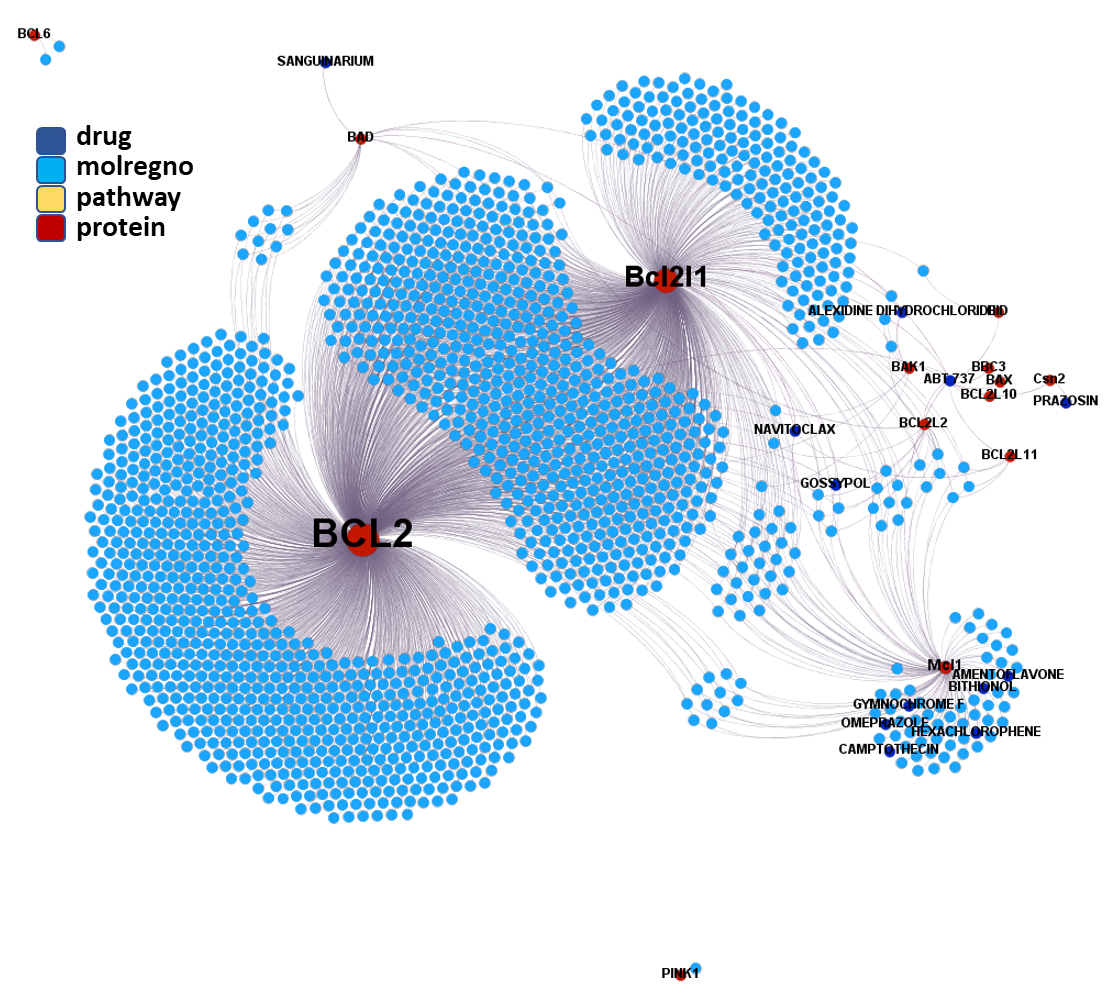
Molecules interacting with BCL2 (B-Cell Lymphoma-2) Apoptosis Regulator

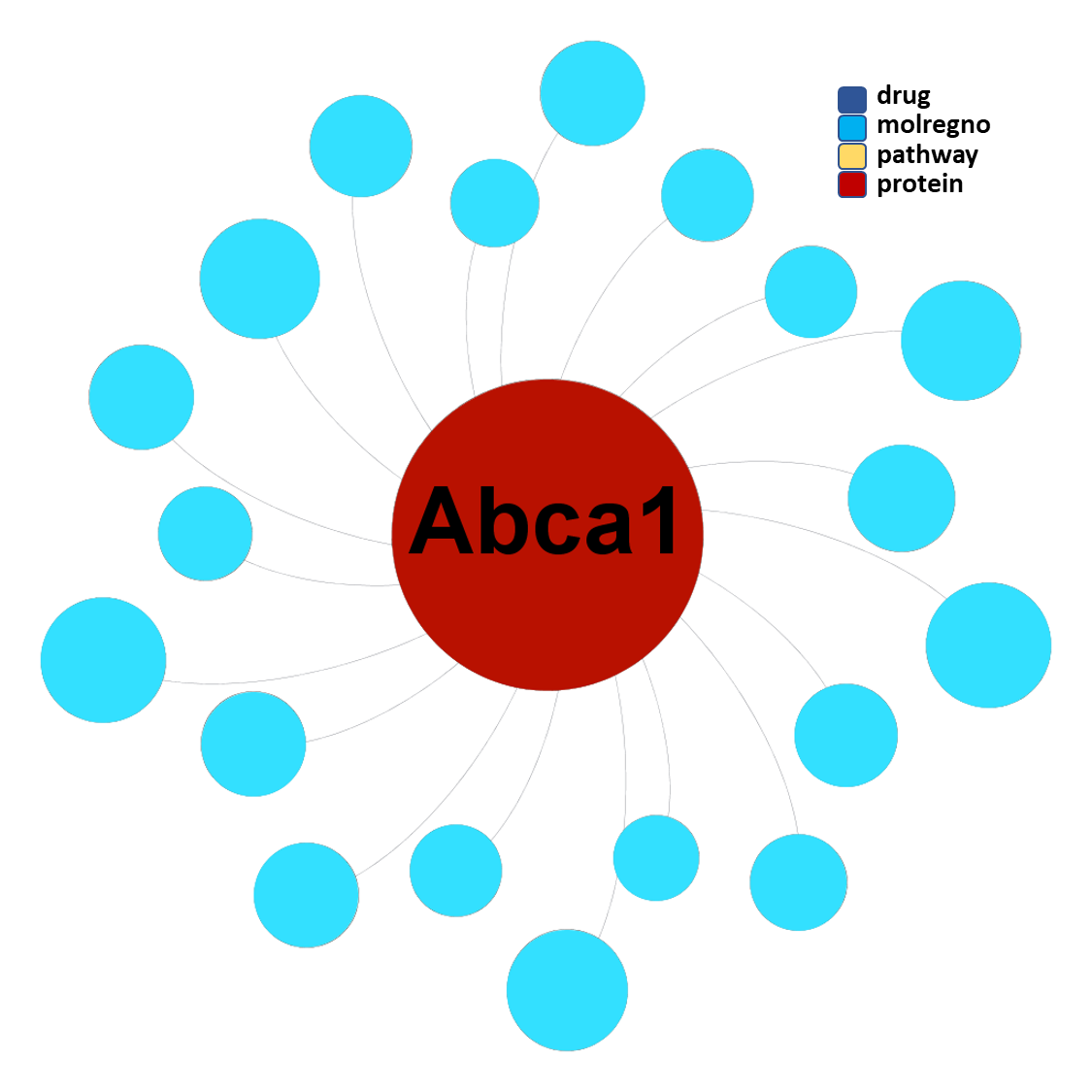
Molecules interacting with Phospholipid-transporting ATPase ABCA1
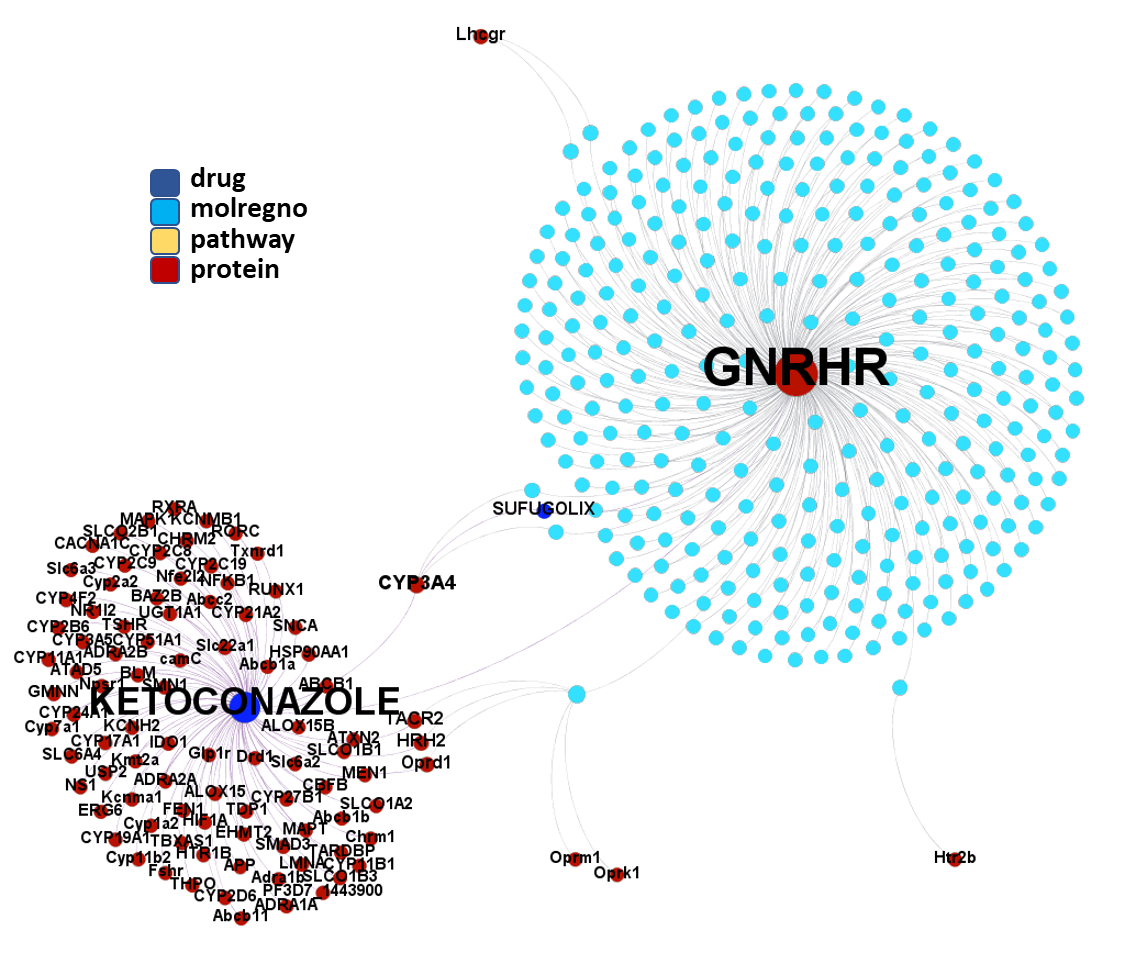
Molecules interacting with Gonadotropin-releasing hormone receptor

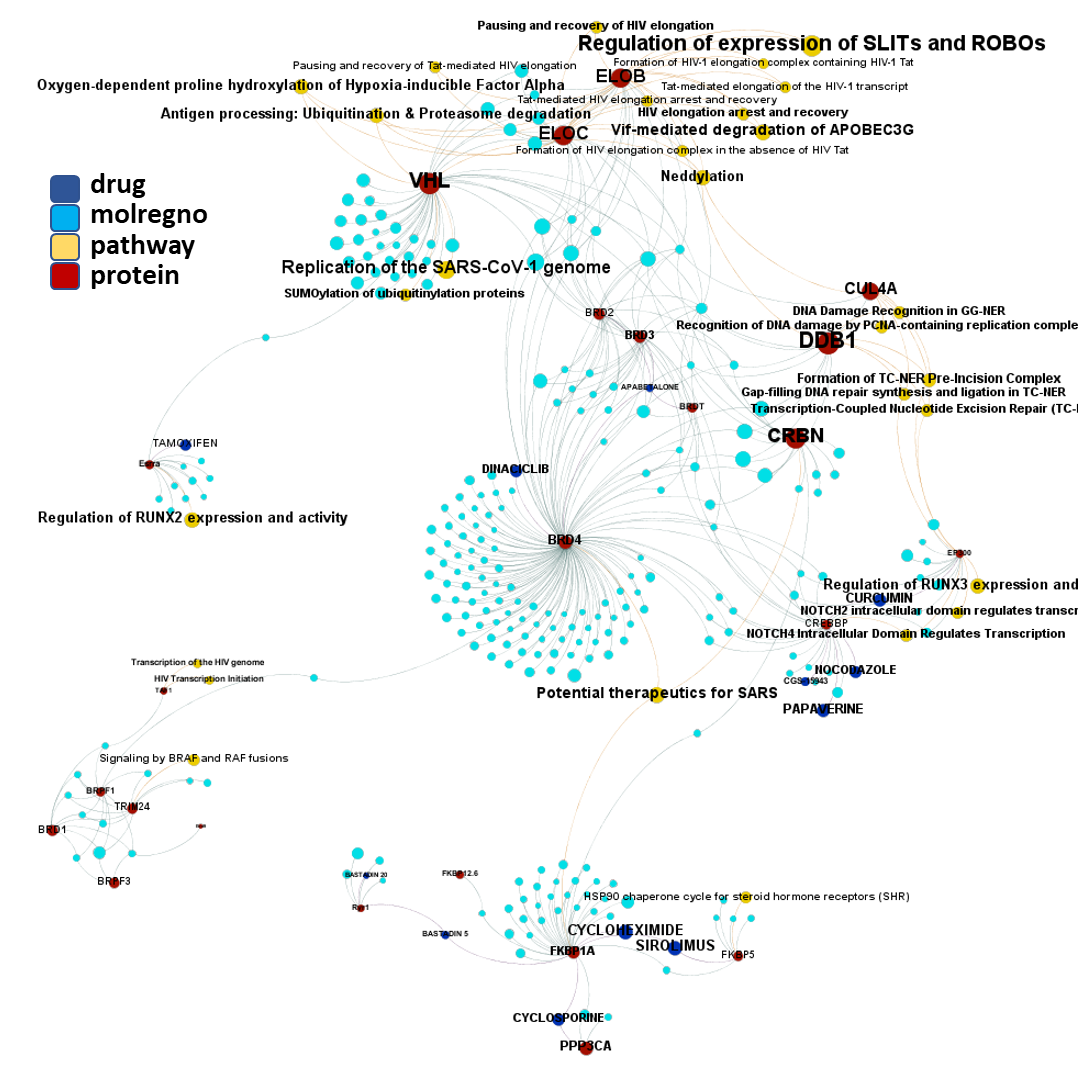
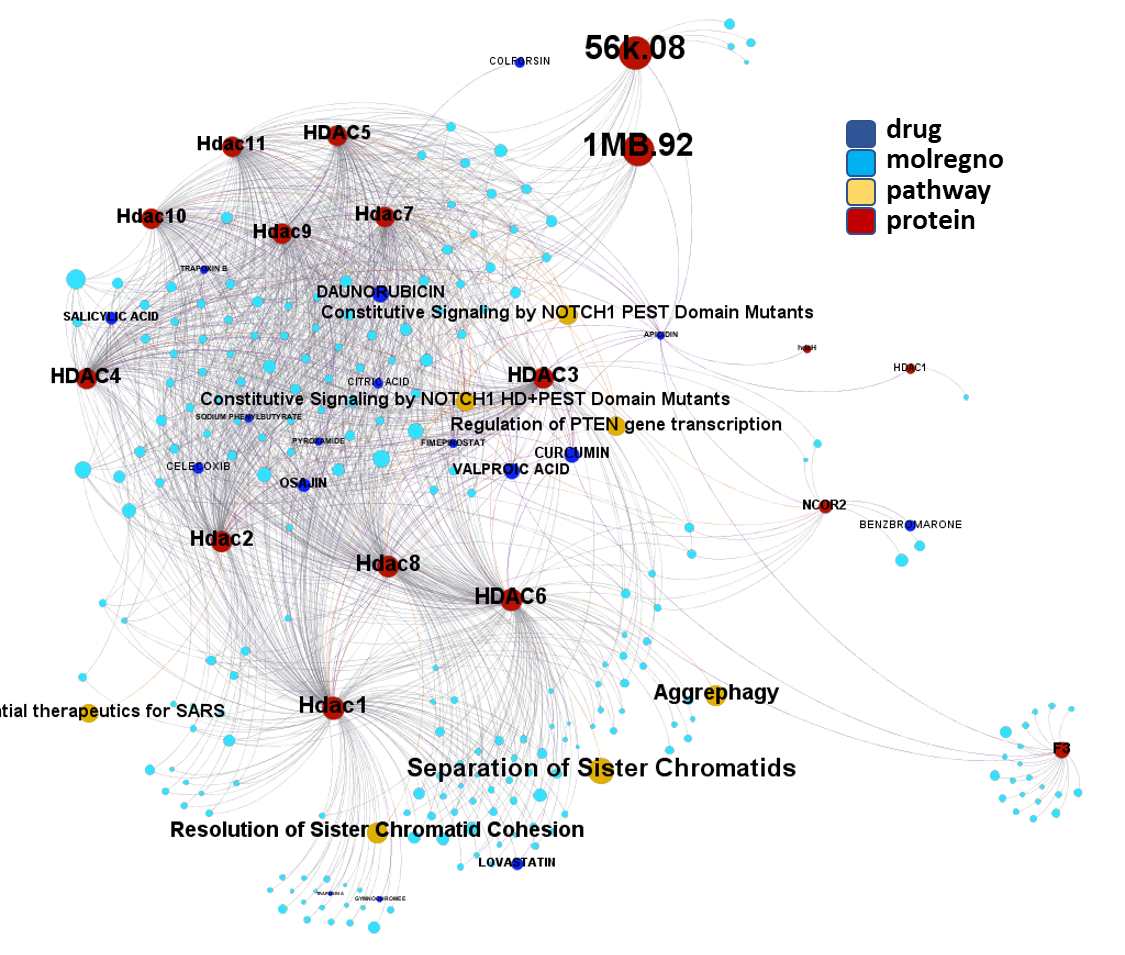
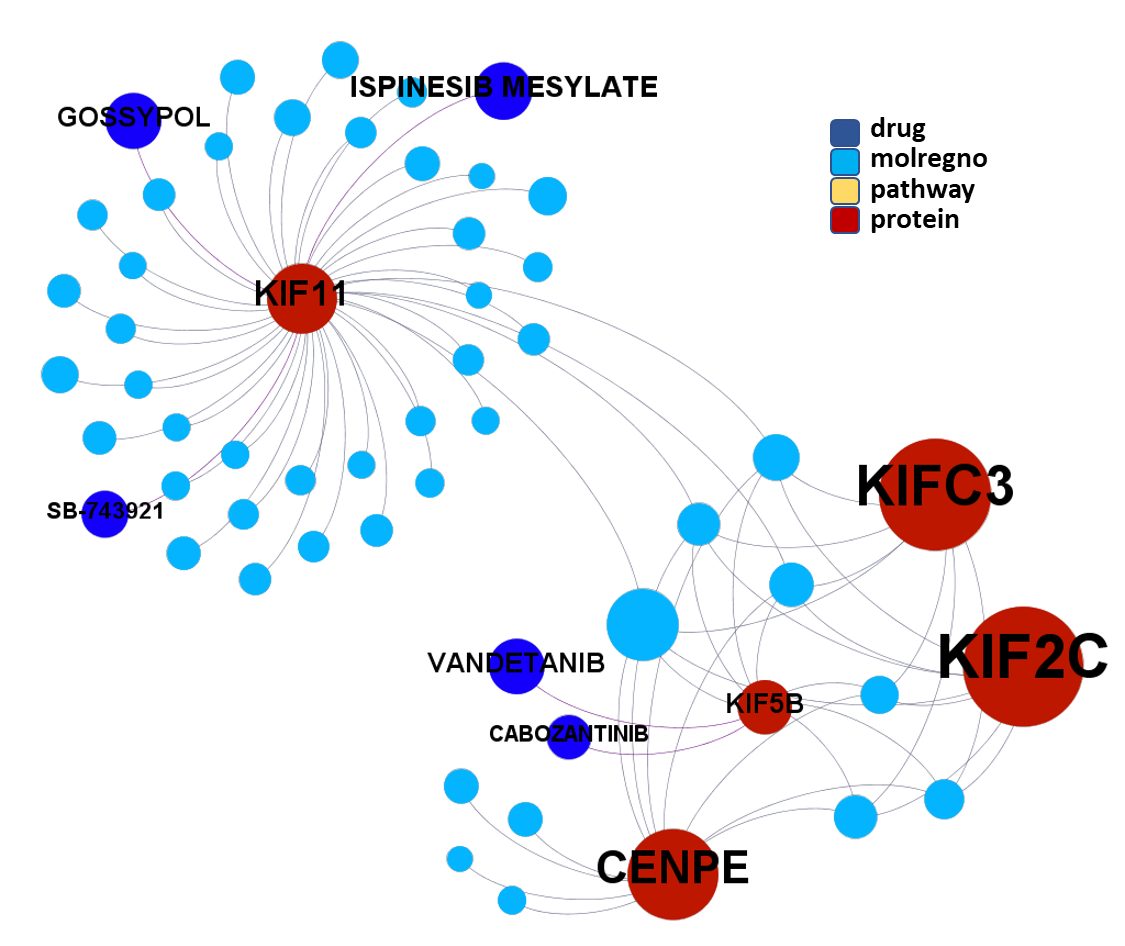
Molecules interacting with Ubiquitin ligases, Bromodomain proteins & FK506-binding proteins