Although no predicted or actual GLP1R PAM has been ranked as > 70% similar to DA-15864 or compound 2, there are about 240 compounds containing the quinoxaline scaffold that both compounds share.
Some exemplars below.
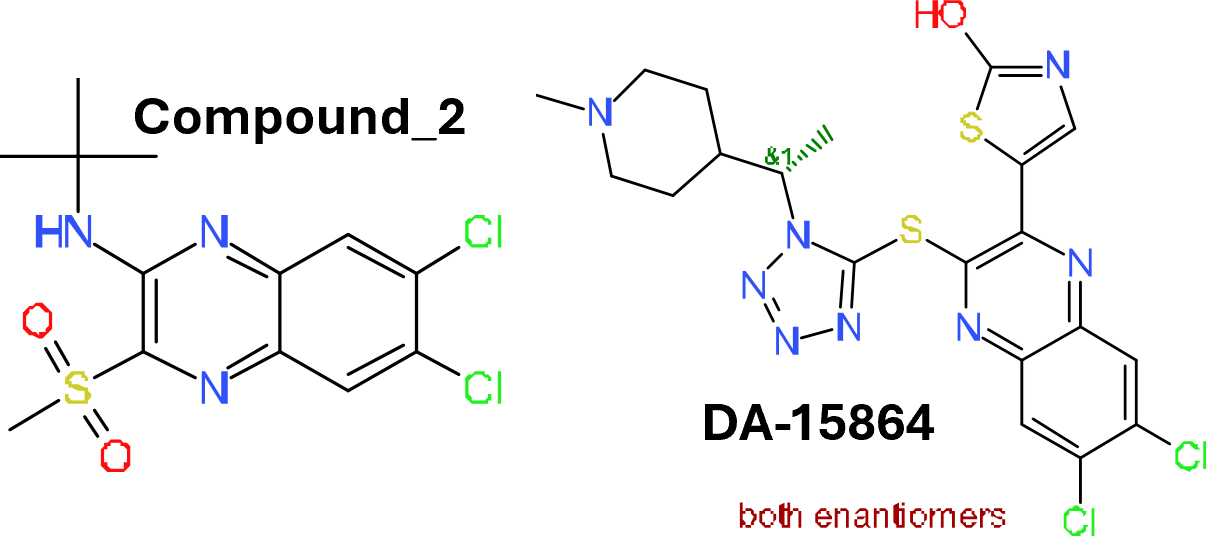
TopLeft: drTargetMolName. Top right: GLP1R PAM score. Down left: parent compound. Down right: whether the score is from experimental values or predicted.
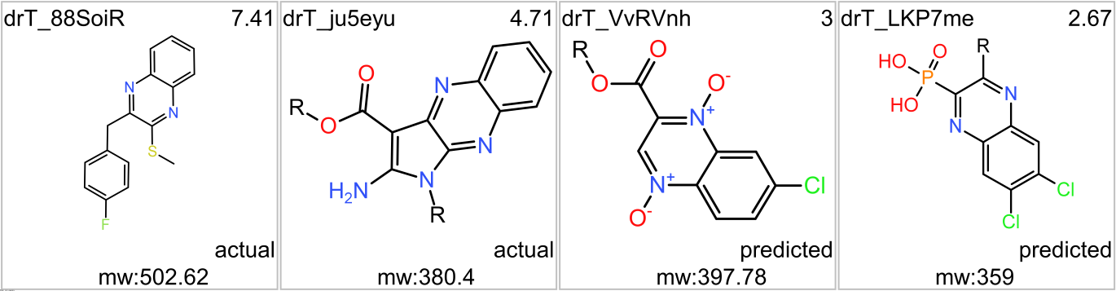
We can use network graps to visualize the interactions of this compounds with relevant ChEMBL phenotypes.
Or check the assays involved in relevant phenotypes

Or assays where some compounds resulted with cytoxic effect

A good number of quinoxalines have been assayed in GPLP1R PChem screens.
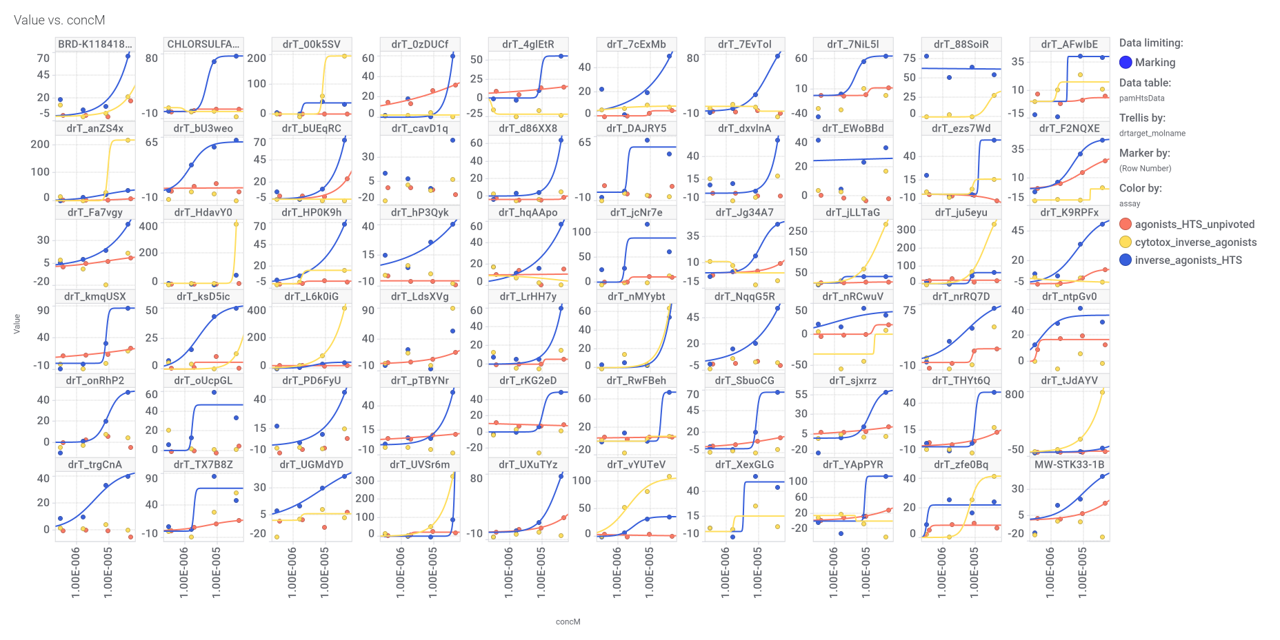
And finally, we can have a look on some of their bio-physicochemical and developability properties.
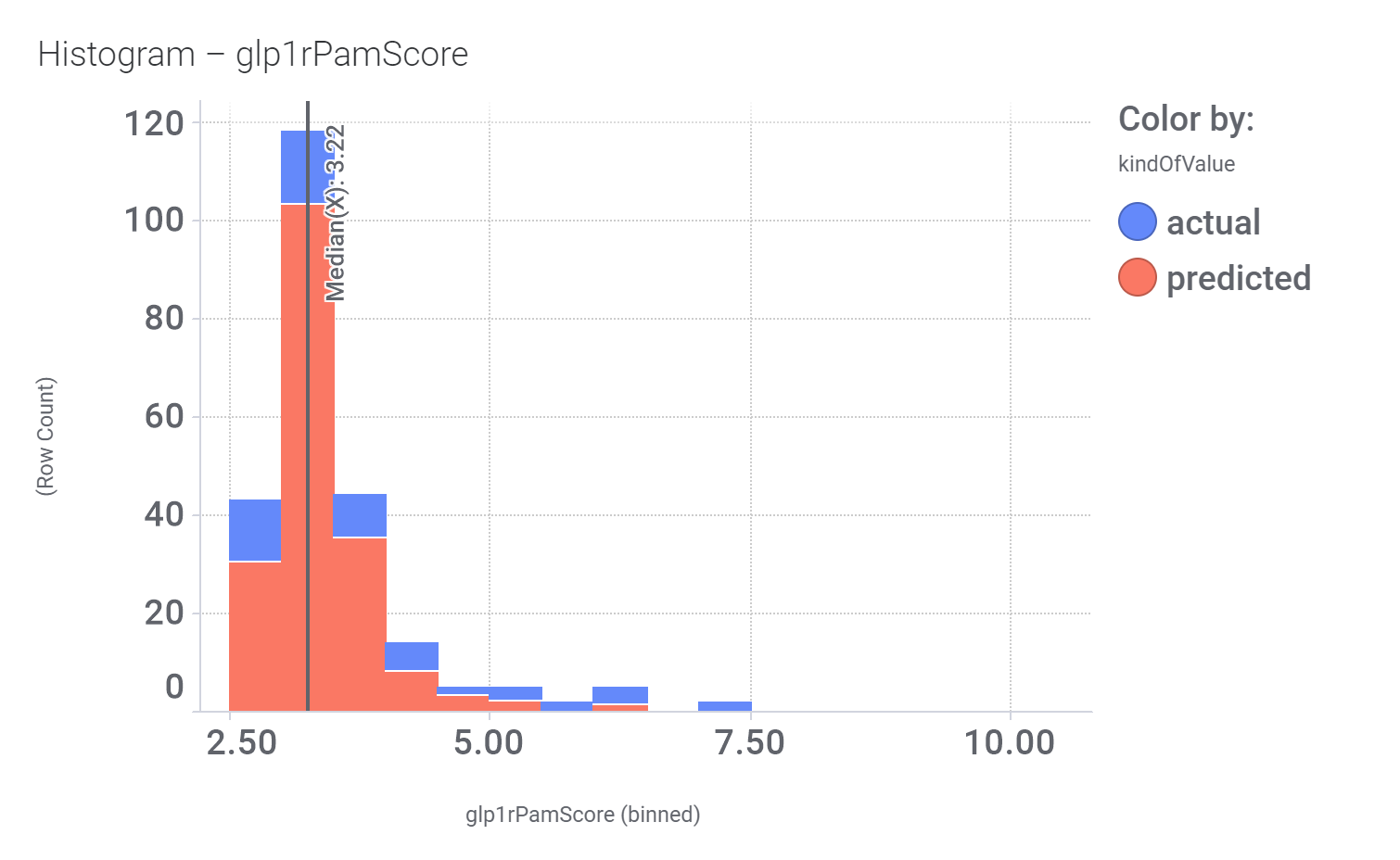
Actual or predicted GLP1R PAM score
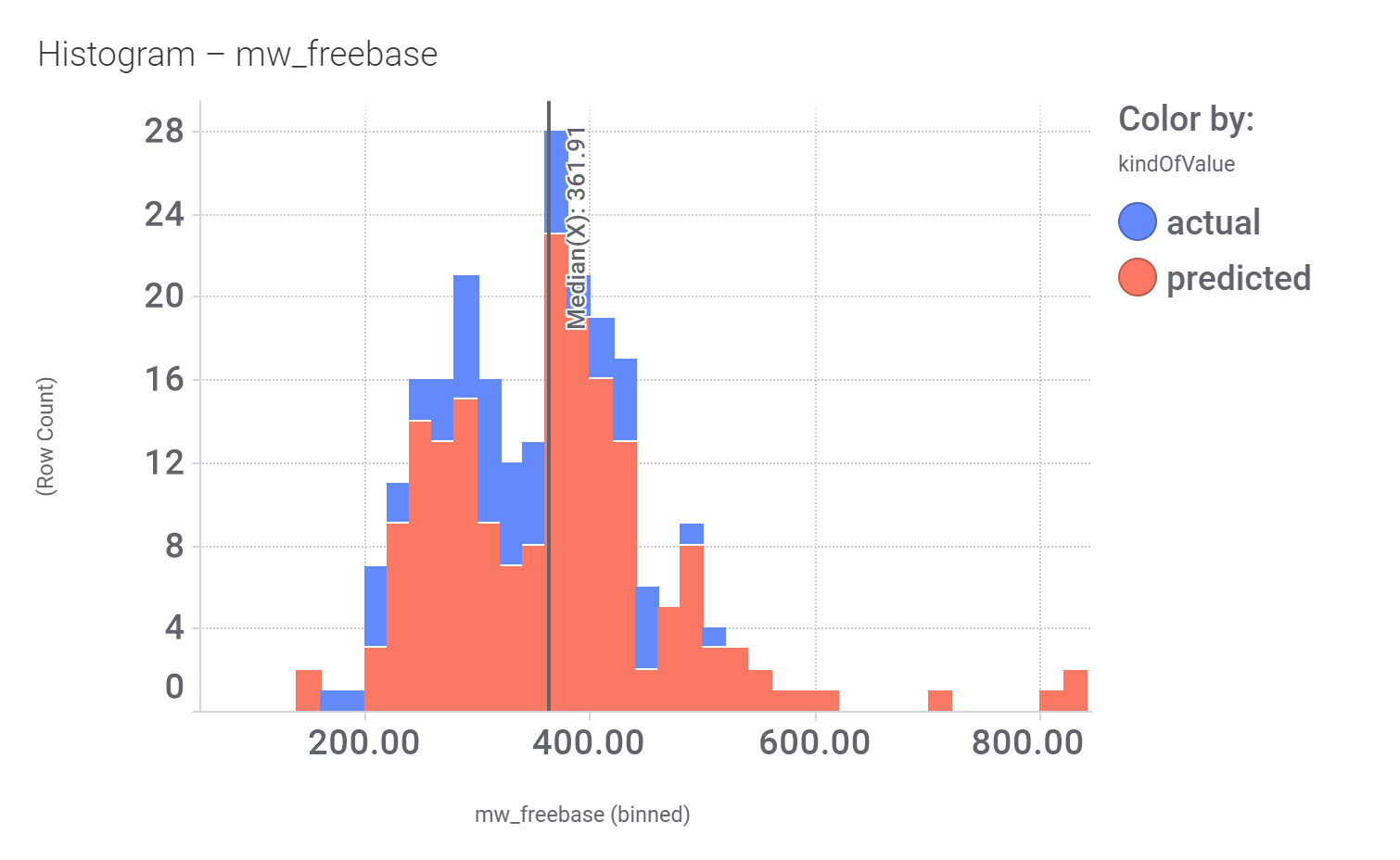
Molecular Weight
Similarity calculated by DataWarrior
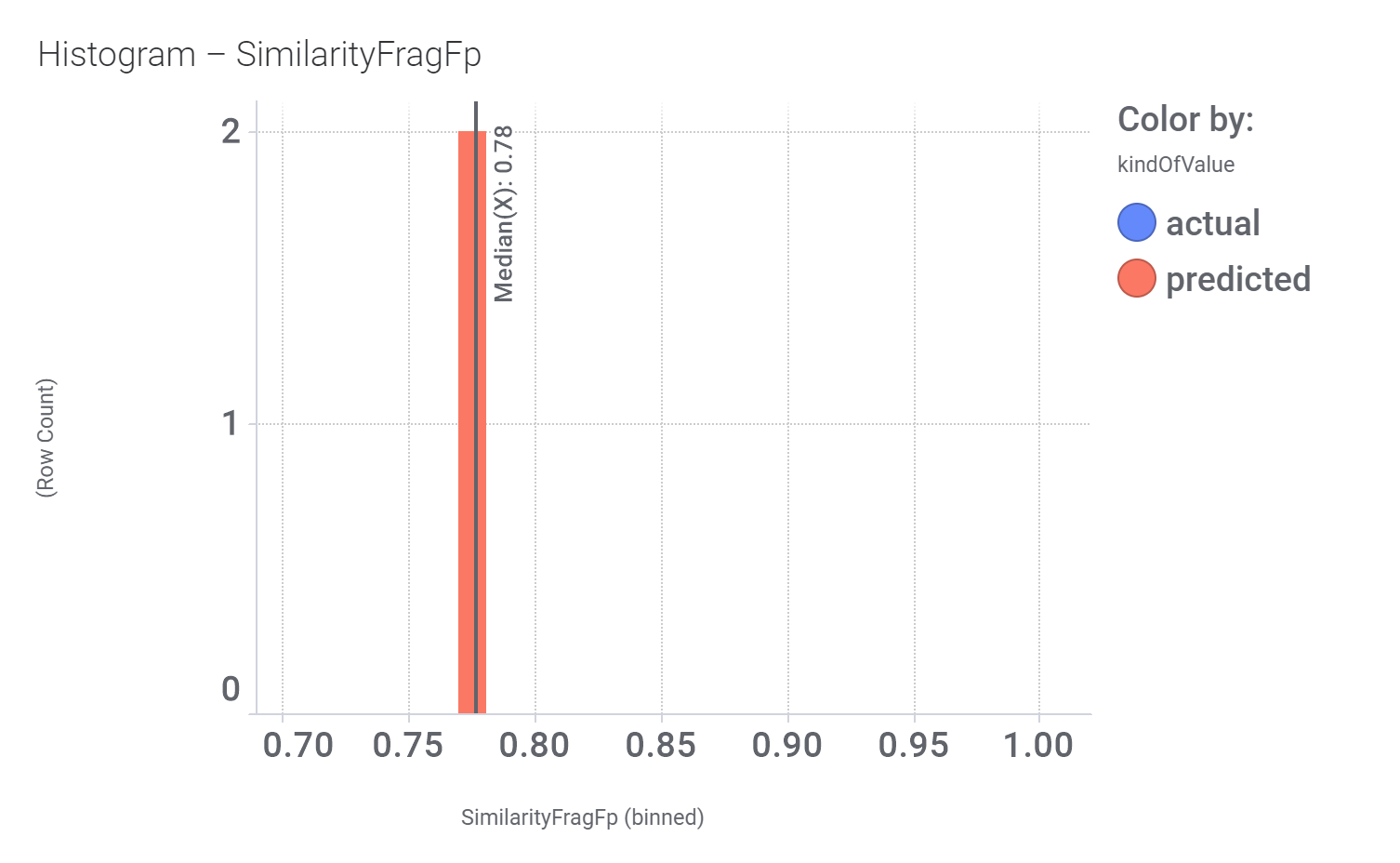
Just 2 quinoxalines showed little similarity with compound_19.
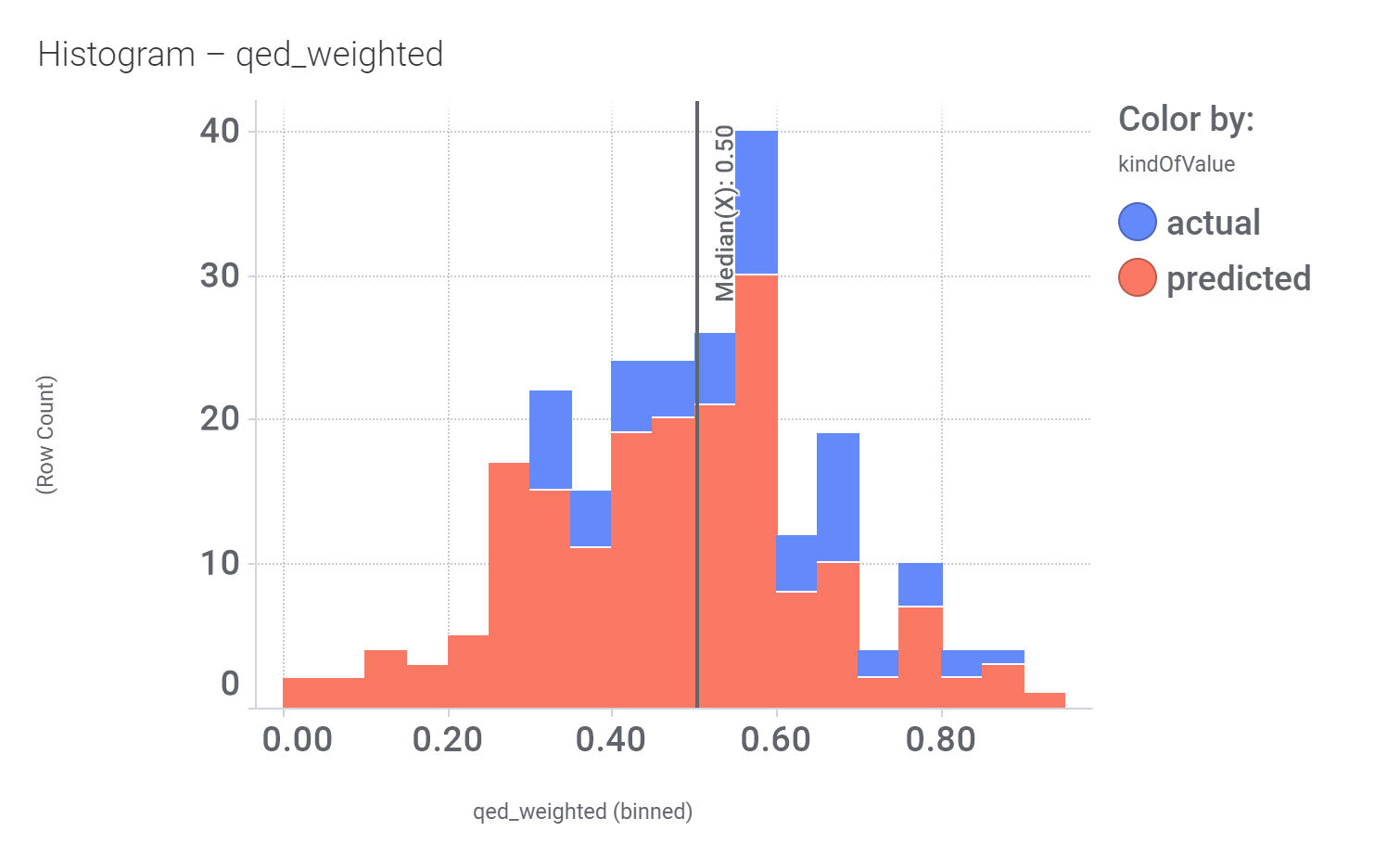
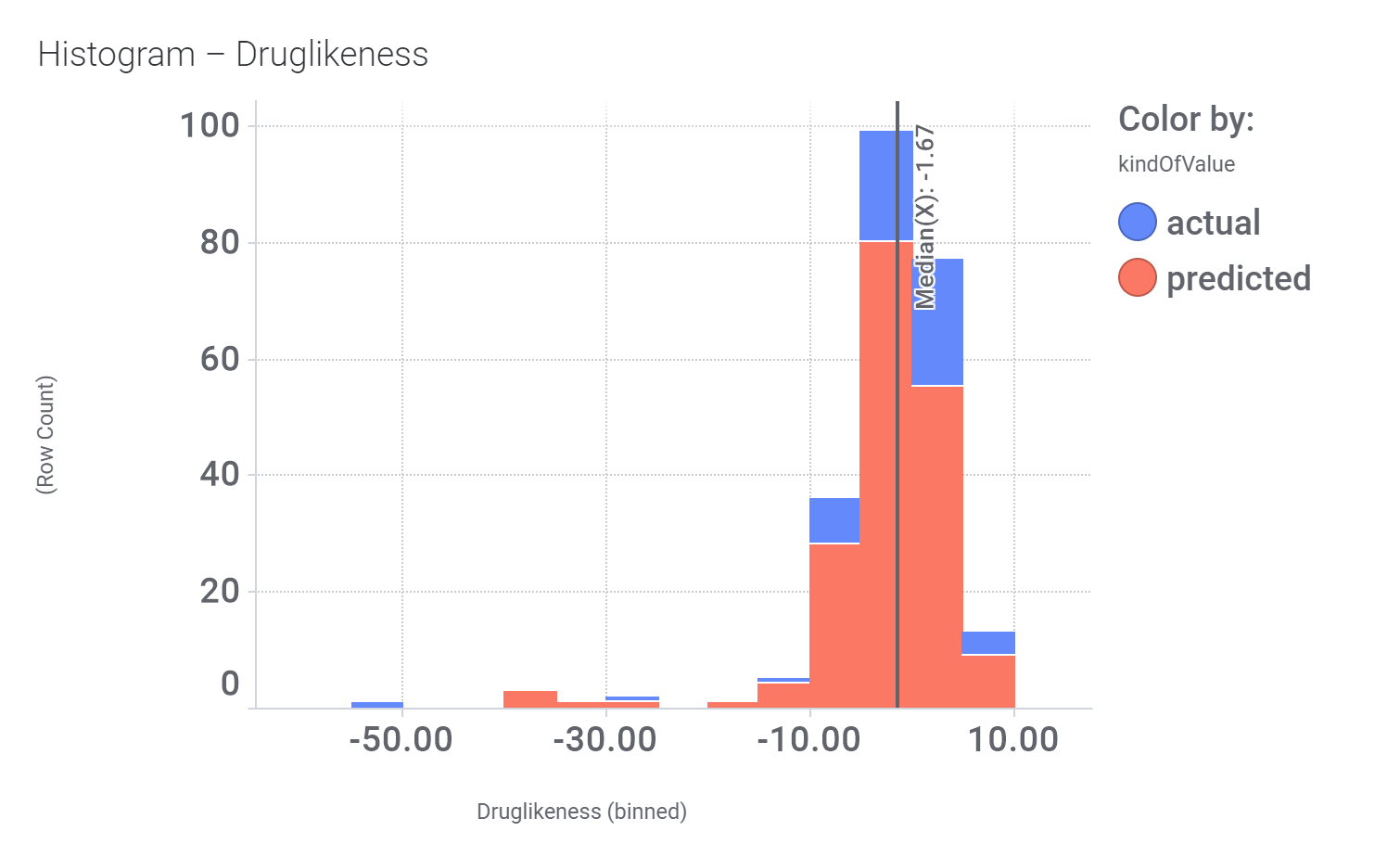
Druglikeness estimated by QedWeighted.
Druglikeness estimated by DataWarrior.
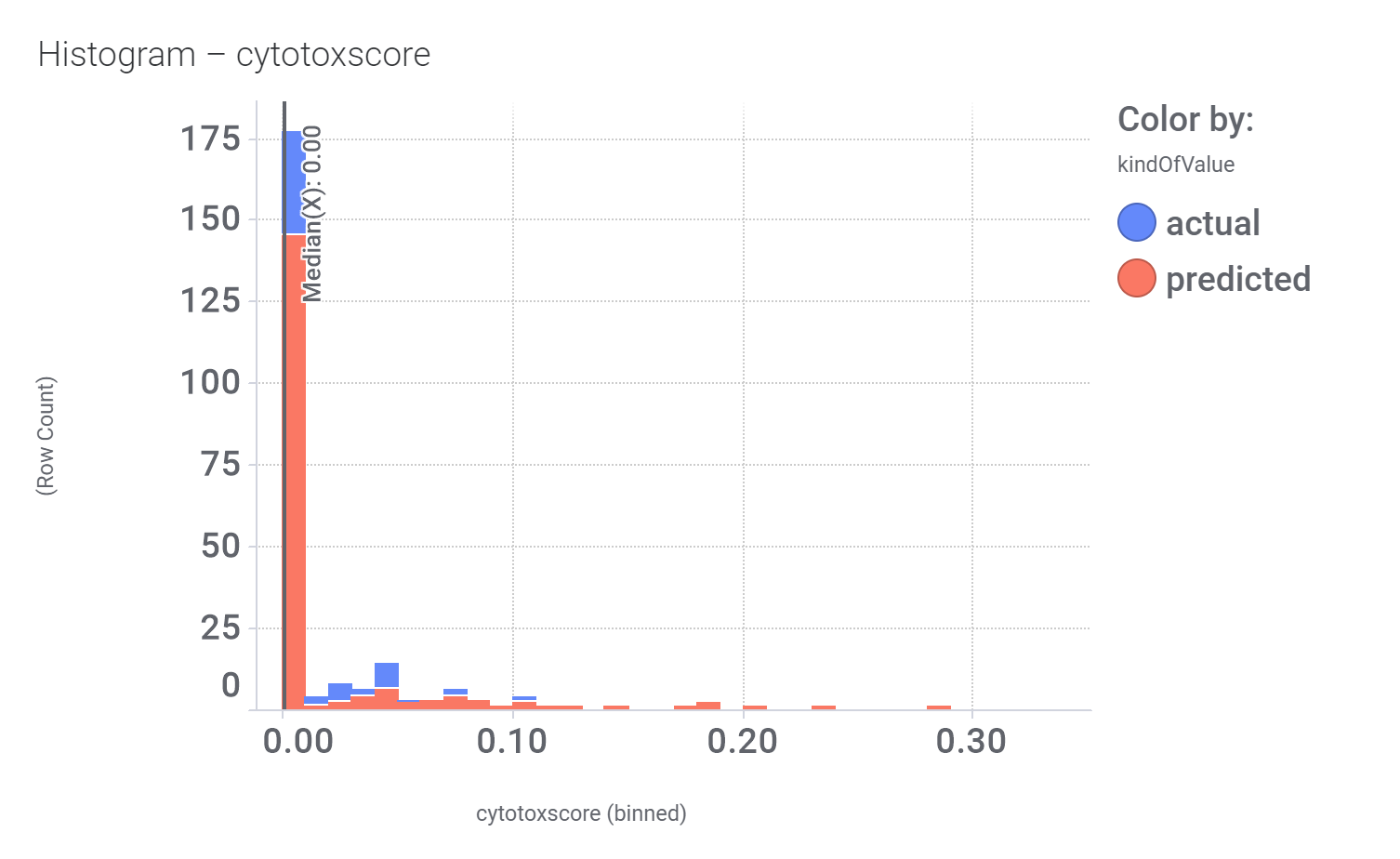
Cytotoxicity score calculated from ChEMBL assays
Literature references.
Br J Pharmacol. 2022 Feb; 179(4): 511–525. doi: 10.1111/bph.15446
Non‐peptide agonists and positive allosteric modulators of glucagon‐like peptide‐1 receptors: Alternative approaches for treatment of Type 2 diabetes
Faisal Malik 1 and Zhijun Li 1
Abstract
Glucagon‐like peptide‐1 (GLP‐1) receptors belong to the pharmaceutically important Class B family of GPCRs and are involved in many biologically significant signalling pathways. Its incretin peptide ligand GLP‐1 analogues are effective treatments for Type 2 diabetes. Although developing non‐peptide low MW drugs targeting GLP‐1 receptors remains elusive, considerable progress has been made in discovering non‐peptide agonists and positive allosteric modulators (PAMs) of GLP‐1 receptors with demonstrated efficacy. Many of these compounds induce biased signalling in GLP‐1 receptor‐mediated functional pathways. High‐quality structures of GLP‐1 receptors in both inactive and active states have been reported, revealing detailed molecular interactions between GLP‐1 receptors and non‐peptide agonists or PAMs. These progresses raise the exciting possibility of developing non‐peptide drugs of GLP‐1 receptors as alternative treatments for Type 2 diabetes. The insight into the interactions between the receptor and the non‐peptide ligand is also useful for developing non‐peptide ligands targeting other Class B GPCRs.
3.1.1. Compound 2 and other quinoxalines
The first reported and also the most extensively studied PAMs of the GLP‐1 receptor are a series of quinoxalines identified by Knudsen et al. (2007) at Novo Nordisk. In a screen for low MW agonists of GLP‐1 receptors, the group first screened 500,000 compounds using a competition‐binding assay and then screened 250,000 compounds using a functional assay, which was followed by structural modifications. This led to the discovery of a series of substituted quinoxalines, as low MW agonists of human GLP‐1 receptors, exemplified by compound 7 (compound 2) (Figure 3, [7]).
Using a cAMP functional assay, compound 2 was shown to be a full agonist of human GLP‐1 receptors and is specific for the human receptor compared with other glucagon B1 GPCRs. Saturation‐binding and functional experiments showed compound 2 to increase the binding affinity of GLP‐1 to GLP‐1 receptors but not its potency. Further, the agonistic effect of compound 2 was not blocked by exendin(9–39). Additional studies determined that compound 2 stimulates glucose‐dependent insulin release from WT mouse islets but not from GLP‐1 receptor knockout mouse islets, and compound 2 and GLP‐1 have additive effects on insulin release. Collectively, these data demonstrate compound 2 interacts with human GLP‐1 receptors at a site different from the orthosteric binding site for GLP‐1 and functions as an agonistic, PAM (ago‐PAM).
The agonistic effect of compound 2 was independently confirmed by a subsequent report from University of Ulster, which examined the in vitro and in vivo metabolic actions of compound 2 (Irwin et al., 2010). Although less potent than GLP‐1, exenatide and liraglutide, compound 2 can significantly stimulate insulin secretion from BRIN‐BD11 cells in a concentration‐dependent fashion. In mice, compound 2 decreases the glucose concentration compared with controls. In another study using a cell line with recombinant expression for the human GLP‐1 receptor and the rat insulinoma cell line INS‐1E expressing GLP‐1 receptors, compound 2 significantly enhanced efficacy and potency of GLP‐1(9–36)NH2 for cAMP accumulation, Ca2+ signalling and pERK1/2, confirming its positive allosteric effects (Li et al., 2012).
With the discovery of compound 2 as an ago‐PAM of GLP‐1 receptors, systematic SAR studies using the quinoxaline scaffold were carried out at its C‐2 position, C‐3 position and the quinoxaline ring (Teng et al., 2007). The most potent and efficacious compounds are found to be 6,7‐dichlo‐roquinoxalines bearing an alkyl sulfonyl group at the C‐2 position and a secondary alkyl amino group at the C‐3 position. However, the active quinoxalines appear chemically unstable when treated with strong nucleophiles, for example, DTT, or strong bases, for example, KOH, and they also have high microsomal turnover rate. Hence, improvement on chemical stability and pharmacokinetic properties of these compounds is necessary before further in vivo characterization is conducted.
Other efforts to explore the quinoxaline scaffold have followed. A series of 2‐thio‐substituted quinoxalines were reported as weak GLP‐1 receptor agonists (Kopin & Beinborn, 2004). Another study reported the synthesis of two series of 3,6,7‐trisubstituted‐2‐(1H‐imidazol‐2‐ylsulfanyl)‐quinoxalines and 2‐(quinoxalin‐2‐yl)‐isothioureas (Bahekar et al., 2007). The glucose‐dependent insulinotropic activity of these compounds was identical to that of compound 2 using a RIN5F cell‐base assay. Moon et al. (2011) disclosed another 2‐thio‐quinoxaline analogue, compound 8 (DA‐15864) (Figure 3, [8]). In CHO cells, DA‐15864 selectively stimulates human with EC50 values of 163 nM and increases intracellular cAMP levels. In rat insulinoma cells, the compound significantly increases glucose‐stimulated insulin secretion and acts synergistically with GLP‐1. In vivo intravenous glucose tolerance tests using normal mice showed that DA‐15864 increased the peak plasma level of insulin significantly. Thus, DA‐15864 appears to be a promising, orally available, GLP‐1 receptor agonist with the potential for treatment of diabetes.
Apart from their location in pancreatic beta‐cells, GLP‐1 receptors are widely expressed in the brain and are also considered to be an effective therapeutic target for diseases of the CNS. Thus, GLP‐1 mimetics including exendin‐4 and liraglutide have neuroprotective effect on ischaemic stroke (Briyal et al., 2012; Darsalia et al., 2014; Li et al., 2009; Sato et al., 2013). Given the agonistic and modulating effect of compound 2 on GLP‐1 receptors, its potential neuroprotective effects on cerebral ischemia were evaluated (H. Zhang et al., 2016). In vitro, compound 2 increased cortical neuron survival in oxygen–glucose deprivation and reperfusion. In middle cerebral artery occlusion (MCAO) mice, compound 2 with exendin‐4 significantly decreased the neurological deficit following MCAO. Using Western blot, GLP‐1 receptors present in cortical neurons could be activated by compound 2. Measuring the stimulated cAMP level in neuronal cells, compound 2 and exendin‐4 induced activation of GLP‐1 receptors, in an additive manner. Therefore, the neuroprotective effects observed are mediated by the activation of the GLP‐1 receptors through the cAMP‐PKA‐CREB signalling pathway.
Proc Natl Acad Sci U S A. 2007 Jan 16; 104(3): 937–942.
Published online 2007 Jan 9. doi: 10.1073/pnas.0605701104. PMID: 17213325
Small-molecule agonists for the glucagon-like peptide 1 receptor
Lotte Bjerre Knudsen,*† Dan Kiel,‡ Min Teng,§ Carsten Behrens,¶ Dilip Bhumralkar,‖ János T. Kodra,¶ Jens J. Holst,** Claus B. Jeppesen,* Michael D. Johnson,§ Johannes Cornelis de Jong,¶ Anker Steen Jorgensen,¶ Tim Kercher,‖ Jarek Kostrowicki,†† Peter Madsen,¶ Preben H. Olesen,¶ Jacob S. Petersen,* Fritz Poulsen,* Ulla G. Sidelmann,‡‡ Jeppe Sturis,§§ Larry Truesdale,‖ John May,‡ and Jesper Lau¶
ABSTRACT
The peptide hormone glucagon-like peptide (GLP)-1 has important actions resulting in glucose lowering along with weight loss in patients with type 2 diabetes. As a peptide hormone, GLP-1 has to be administered by injection. Only a few small-molecule agonists to peptide hormone receptors have been described and none in the B family of the G protein coupled receptors to which the GLP-1 receptor belongs. We have discovered a series of small molecules known as ago-allosteric modulators selective for the human GLP-1 receptor. These compounds act as both allosteric activators of the receptor and independent agonists. Potency of GLP-1 was not changed by the allosteric agonists, but affinity of GLP-1 for the receptor was increased. The most potent compound identified stimulates glucose-dependent insulin release from normal mouse islets but, importantly, not from GLP-1 receptor knockout mice. Also, the compound stimulates insulin release from perfused rat pancreas in a manner additive with GLP-1 itself. These compounds may lead to the identification or design of orally active GLP-1 agonists.
Eur J Pharmacol. 2010 Feb 25;628(1-3):268-73.
DOI: 10.1016/j.ejphar.2009.11.022. PMID: 19917278
Insulin-releasing and metabolic effects of small molecule GLP-1 receptor agonist 6,7-dichloro-2-methylsulfonyl-3-N-tert-butylaminoquinoxaline
Nigel Irwin 1, Peter R Flatt, Steven Patterson, Brian D Green
Abstract
Much recent attention has focused on the GLP-1 receptor as a potential target for antidiabetic drugs. Enzyme resistant GLP-1 mimetics such as exenatide are now employed for the treatment of type 2 diabetes, but must be administered by injection. The present study has examined and compared the in vitro and in vivo metabolic actions of a small molecule GLP-1 receptor agonist 6,7-dichloro-2-methylsulfonyl-3-N-tert-butylaminoquinoxaline (DMB), with native GLP-1, exenatide and liraglutide. DMB significantly stimulated in vitro insulin secretion from BRIN-BD11 cells but with decreased molar potency compared to native GLP-1 or related mimetics. Administration of DMB in combination with glucose to mice significantly (P<0.05) decreased the overall glucose excursion compared to controls. Exenatide and liraglutide evoked similar (P<0.001) reductions of the overall glycaemic excursion, but were significantly (P<0.001 and P<0.05; respectively) more effective than DMB. These observations were associated with prominently (P<0.05) enhanced glucose-mediated insulin release by exenatide and liraglutide, but not by DMB. Combined injection of DMB with either liraglutide or exenatide did not substantially improve glucose-lowering or insulin-releasing responses. However, administration of DMB in combination with exendin(9-39) did not impair its glucoregulatory actions. These results provide evidence to support the development and potential use of low molecular weight GLP-1 receptor agonists for the treatment of type 2 diabetes.
PLoS One. 2012; 7(10): e47936.
Published online 2012 Oct 19. doi: 10.1371/journal.pone.0047936
PMCID: PMC3477139
PMID: 23094100
Allosteric Modulation of the Activity of the Glucagon-like Peptide-1 (GLP-1) Metabolite GLP-1 9–36 Amide at the GLP-1 Receptor
Naichang Li, 1 , 2 Jing Lu, 1 and Gary B. Willars 1 , *
Alexander G. Obukhov, Editor
Abstract
Glucagon-like peptide-1 (GLP-1) released from intestinal L cells in response to nutrients has many physiological effects but particularly enhances glucose-dependent insulin release through the GLP-1 receptor (GLP-1R). GLP-1 7–36 amide, the predominant circulating active form of GLP-1, is rapidly truncated by dipeptidyl peptidase-4 to GLP-1 9–36 amide, which is generally considered inactive. Given its physiological roles, the GLP-1R is targeted for treatment of type 2 diabetes. Recently ‘compound 2’ has been described as both an agonist and positive allosteric modulator of GLP-1 7–36 amide affinity, but not potency, at the GLP-1R. Importantly, we demonstrated previously that exendin 9–39, generally considered a GLP-1R antagonist, enhances compound 2 efficacy (or vice versa) at the GLP-1R. Given that GLP-1 9–36 amide is the major circulating form of GLP-1 post-prandially and is a low affinity weak partial agonist or antagonist at the GLP-1R, we investigated interaction between this metabolite and compound 2 in a cell line with recombinant expression of the human GLP-1R and the rat insulinoma cell line, INS-1E, with native expression of the GLP-1R. We show compound 2 markedly enhances efficacy and potency of GLP-1 9–36 amide for key cellular responses including AMP generation, Ca2+ signaling and extracellular signal-regulated kinase. Thus, metabolites of peptide hormones including GLP-1 that are often considered inactive may provide a means of manipulating key aspects of receptor function and a novel therapeutic strategy.
Bioorg Med Chem Lett. 2007 Oct 1;17(19):5472-8.
DOI: 10.1016/j.bmcl.2007.06.086. PMID: 17827014
Small molecule ago-allosteric modulators of the human glucagon-like peptide-1 (hGLP-1) receptor
Min Teng 1, Michael D Johnson, Christine Thomas, Dan Kiel, James N Lakis, Tim Kercher, Shelley Aytes, Jarek Kostrowicki, Dilip Bhumralkar, Larry Truesdale, John May, Ulla Sidelman, Janos T Kodra, Anker Steen Jørgensen, Preben Houlberg Olesen, Johannes Cornelis de Jong, Peter Madsen, Carsten Behrens, Ingrid Pettersson, Lotte Bjerre Knudsen, Jens J Holst, Jesper Lau
Abstract
Following our previous publication describing the biological profiles, we herein describe the structure-activity relationships of a core set of quinoxalines as the hGLP-1 receptor agonists. The most potent and efficacious compounds are 6,7-dichloroquinoxalines bearing an alkyl sulfonyl group at the C-2 position and a secondary alkyl amino group at the C-3 position. These findings serve as a valuable starting point for the discovery of more drug-like small molecule agonists for the hGLP-1 receptor.
- Kopin, A. , & Beinborn, M. (2004). Methods and compositions for the treatment of metabolic disorders. WO/2004/103310.
Arch Pharm (Weinheim). 2007 Jul;340(7):359-66.
DOI: 10.1002/ardp.200700024. PMID: 17567824
Synthesis and antidiabetic activity of 3,6,7-trisubstituted-2-(1H-imidazol-2-ylsulfanyl)quinoxalines and quinoxalin-2-yl isothioureas
Rajesh H Bahekar 1, Mukul R Jain, Arun A Gupta, Ashish Goel, Pradip A Jadav, Dipam N Patel, Vijay M Prajapati, Pankaj R Patel
Abstract
Two series of 3,6,7-trisubstituted-2-(1H-imidazol-2-ylsulfanyl)-quinoxalines 2a-l and 2-(quinoxalin-2-yl)-isothioureas 3a-l were prepared. All the test compounds 2a-l and 3a-l were screened in vitro, in a RIN5F cell-based assay for glucose-dependent insulinotropic activity. A significant concentration and glucose-dependent insulin secretion effect was seen with compounds 2a-l and the insulinotropic activity of compound 2l was found to be identical to that of the standard compound (6,7-dichloro-2-trifluromethyl-3-(5-methyl-1,3,4-thiadiazo-2-ylsulfanyl)-quinoxaline (1)).
PLoS One. 2016; 11(2): e0148827. Published online 2016 Feb 10.
doi: 10.1371/journal.pone.0148827. PMCID: PMC4749391. PMID: 26863436
An Orally Active Allosteric GLP-1 Receptor Agonist Is Neuroprotective in Cellular and Rodent Models of Stroke
Huinan Zhang,#1 Yunhan Liu,#2 Shaoyu Guan,#1 Di Qu,1 Ling Wang,3 Xinshang Wang,1 Xubo Li,1 Shimeng Zhou,1 Ying Zhou,1 Ning Wang,1 Jingru Meng,1,* and Xue Ma1,*
Abstract
Diabetes is a major risk factor for the development of stroke. Glucagon-like peptide-1 receptor (GLP-1R) agonists have been in clinical use for the treatment of diabetes and also been reported to be neuroprotective in ischemic stroke. The quinoxaline 6,7-dichloro-2-methylsulfonyl-3-N-tert- butylaminoquinoxaline (DMB) is an agonist and allosteric modulator of the GLP-1R with the potential to increase the affinity of GLP-1 for its receptor. The aim of this study was to evaluate the neuroprotective effects of DMB on transient focal cerebral ischemia. In cultured cortical neurons, DMB activated the GLP-1R, leading to increased intracellular cAMP levels with an EC50 value about 100 fold that of exendin-4. Pretreatment of neurons with DMB protected against necrotic and apoptotic cell death was induced by oxygen-glucose deprivation (OGD). The neuroprotective effects of DMB were blocked by GLP-1R knockdown with shRNA but not by GLP-1R antagonism. In C57BL/6 mice, DMB was orally administered 30 min prior to middle cerebral artery occlusion (MCAO) surgery. DMB markedly reduced the cerebral infarct size and neurological deficits caused by MCAO and reperfusion. The neuroprotective effects were mediated by activation of the GLP-1R through the cAMP-PKA-CREB signaling pathway. DMB exhibited anti-apoptotic effects by modulating Bcl-2 family members. These results provide evidence that DMB, a small molecular GLP-1R agonist, attenuates transient focal cerebral ischemia injury and inhibits neuronal apoptosis induced by MCAO. Taken together, these data suggest that DMB is a potential neuroprotective agent against cerebral ischemia.